|
Special Issue:
SPECIAL TOPIC — Modeling and simulations for the structures and functions of proteins and nucleic acids
|
| TOPICAL REVIEW—Modeling and simulations for the structures and functions of proteins and nucleic acids |
Prev
Next
|
|
|
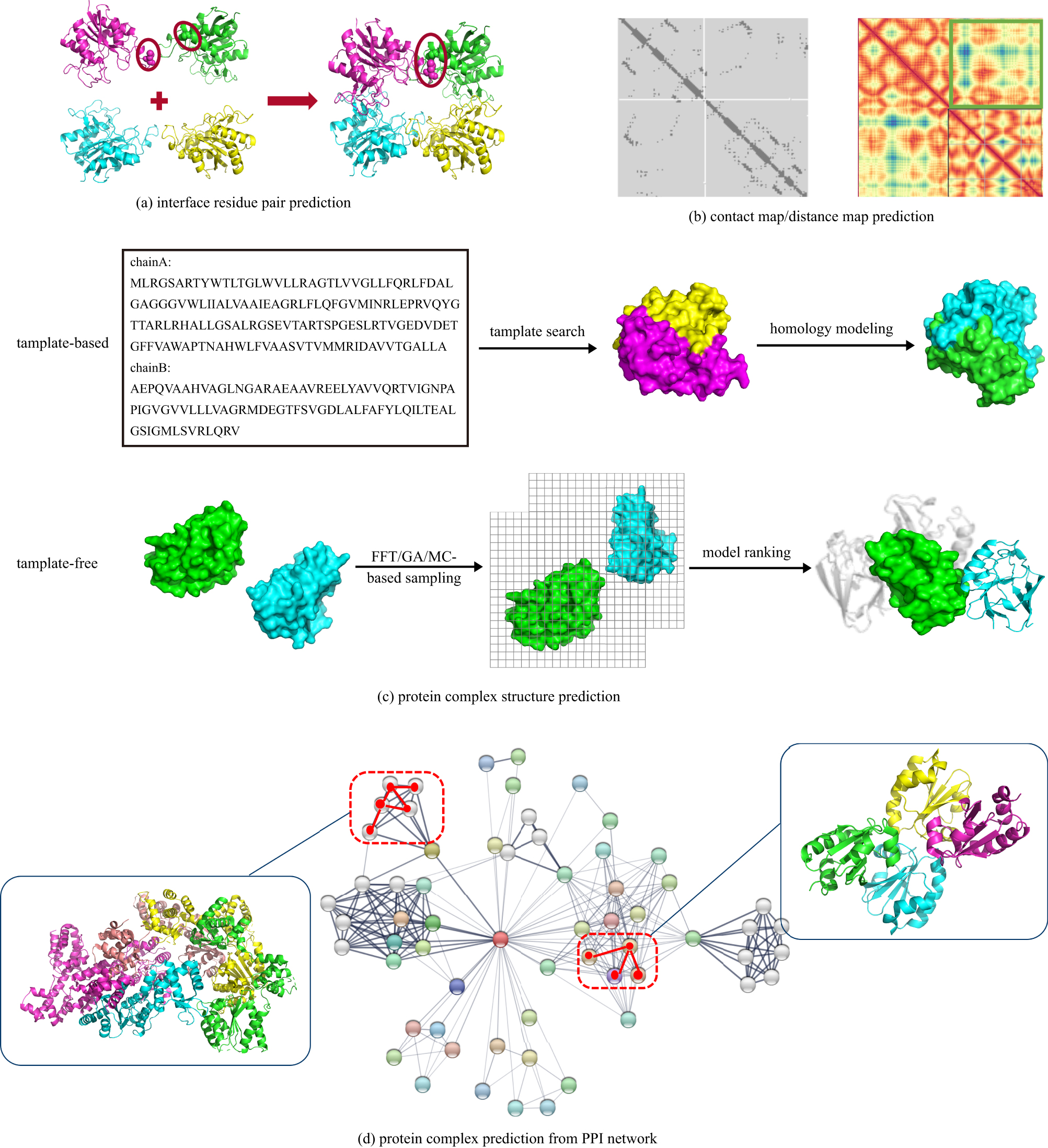
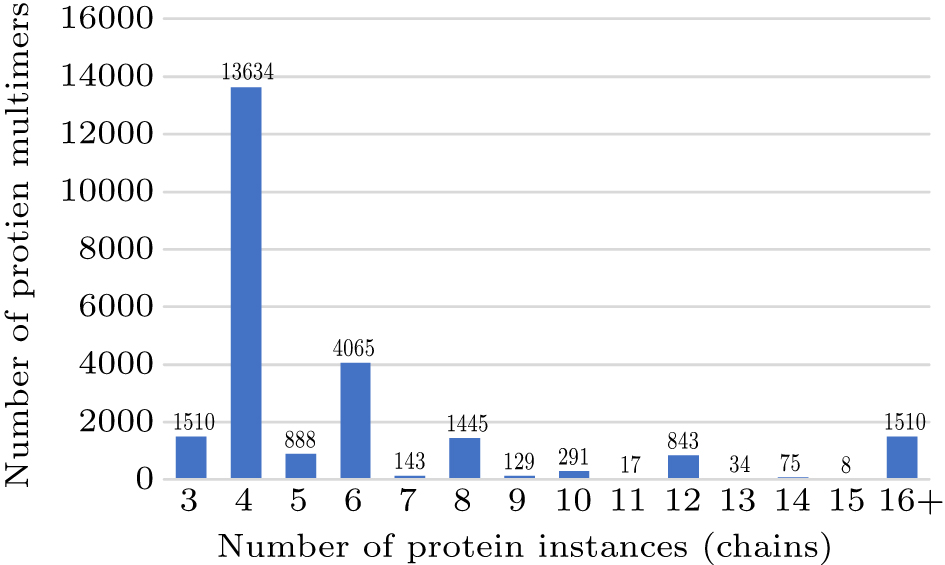
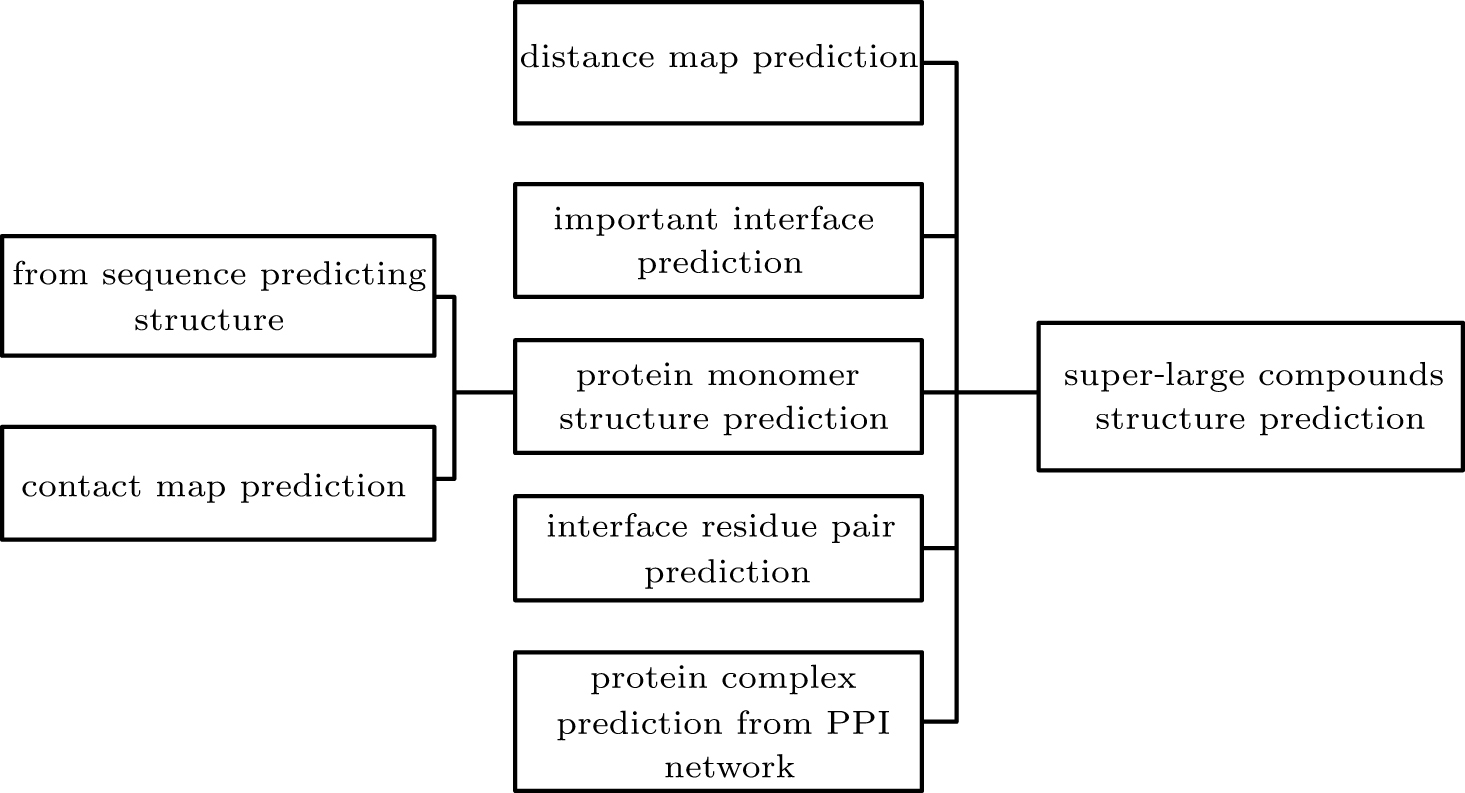
Review of multimer protein–protein interaction complex topology and structure prediction |
| Daiwen Sun(孙黛雯)1, Shijie Liu(刘世婕)1, and Xinqi Gong(龚新奇)1,2,† |
1 Mathematics Intelligence Application Laboratory, Institute for Mathematical Sciences, Renmin University of China, Beijing 100872, China
2 Beijing Advanced Innovation Center for Structural Biology, Tshinghua University, Beijing 100094, China |
|
|
|
|
Abstract Protein–protein interactions (PPI) are important for many biological processes. Theoretical understanding of the structurally determining factors of interaction sites will help to understand the underlying mechanism of protein–protein interactions. At the same time, understanding the complex structure of proteins helps to explore their function. And accurately predicting protein complexes from PPI networks helps us understand the relationship between proteins. In the past few decades, scholars have proposed many methods for predicting protein interactions and protein complex structures. In this review, we first briefly introduce the methods and servers for predicting protein interaction sites and interface residue pairs, and then introduce the protein complex structure prediction methods including template-based prediction and template-free prediction. Subsequently, this paper introduces the methods of predicting protein complexes from the PPI network and the method of predicting missing links in the PPI network. Finally, it briefly summarizes the application of machine/deep learning models in protein structure prediction and action site prediction.
|
Received: 29 June 2020
Revised: 31 August 2020
Accepted manuscript online: 09 September 2020
|
|
PACS:
|
87.15.km
|
(Protein-protein interactions)
|
| |
01.50.hv
|
(Computer software and software reviews)
|
|
|
Corresponding Authors:
†Corresponding author. E-mail: xinqigong@ruc.edu.cn
|
| About author: †Corresponding author. E-mail: xinqigong@ruc.edu.cn * Project supported by the National Natural Science Foundation of China (Grant No. 31670725). |
Cite this article:
Daiwen Sun(孙黛雯), Shijie Liu(刘世婕), and Xinqi Gong(龚新奇)† Review of multimer protein–protein interaction complex topology and structure prediction 2020 Chin. Phys. B 29 108707
|
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
Zeng H, Wang S, Zhou T, Zhao F, Li X, Wu Q, Xu J 2018 Nucleic Acids Research 46 W432 DOI: 10.1093/nar/gky420 |
| [5] |
Ching T, Himmelstein D S, Beaulieu-Jones B K et al. 2018 J. R. Soc. Interface 15 20170387 DOI: 10.1098/rsif.2017.0387 |
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
Balakrishnan S, Kamisetty H, Carbonell J G, Lee S I, Langmead C J 2011 Proteins 79 1061 DOI: 10.1002/prot.22934 |
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
Wang S, Li W, Zhang R, Liu S, Xu J 2016 Nucleic Acids Res. 44 W361 DOI: 10.1093/nar/gkw307 |
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
Yan Y, Zhang D, Zhou P, Li B, Huang S Y 2017 Nucleic Acids Res. 45 W365 DOI: 10.1093/nar/gkx407 |
| [30] |
|
| [31] |
|
| [32] |
Yu H, Luscombe N M, Lu H X, Zhu X, Xia Y, Han J D J, Bertin N, Chung S, Vidal M, Gerstein M 2004 Genome Research 14 1107 DOI: 10.1101/gr.1774904 |
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
Aloy P, Böttcher B, Ceulemans H, Leutwein C, Mellwig C, Fischer S, Gavin A C, Bork P, Superti-Furga G, Serrano L 2004 Science 303 2026 DOI: 10.1126/science.1092645 |
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
Yu H, Braun P, Yildirim M A, Lemmens I, Venkatesan K, Sahalie J, Hirozane-Kishikawa T, Gebreab F, Li N, Simonis N, Hao T, Rual J F, Dricot A, Vazquez A, Murray R R, Simon C, Tardivo L, Tam S, Svrzikapa N, Fan C, de Smet A S, Motyl A, Hudson M E, Park J, Xin X, Cusick M E, Moore T, Boone C, Snyder M, Roth F P, Barabási A L, Tavernier J, Hill D E, Vidal M 2008 Science 322 104 DOI: 10.1126/science.1158684 |
| [43] |
Tarassov K, Messier V, Landry C R, Radinovic S, Serna Molina M M, Shames I, Malitskaya Y, Vogel J, Bussey H, Michnick S W 2008 Science 320 1465 DOI: 10.1126/science.1153878 |
| [44] |
|
| [45] |
Gavin A C, Aloy P, Grandi P, Krause R, Boesche M, Marzioch M, Rau C, Jensen L J, Bastuck S, Dümpelfeld B, Edelmann A, Heurtier M A, Hoffman V, Hoefert C, Klein K, Hudak M, Michon A M, Schelder M, Schirle M, Remor M, Rudi T, Hooper S, Bauer A, Bouwmeester T, Casari G, Drewes G, Neubauer G, Rick J M, Kuster B, Bork P, Russell R B, Superti-Furga G 2006 Nature 440 631 DOI: 10.1038/nature04532 |
| [46] |
|
| [47] |
|
| [48] |
Jeong H, Mason S P, Barabási A L, Oltvai Z N 2001 Nature 411 41 DOI: 10.1038/35075138 |
| [49] |
Han J D J, Bertin N, Hao T, Goldberg D S, Berriz G F, Zhang L V, Dupuy D, Walhout A J M, Cusick M E, Roth F P, Vidal M 2004 Nature 430 88 DOI: 10.1038/nature02555 |
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
Asthana S, King O D, Gibbons F D, Roth F P 2004 Genome Research 14 1170 DOI: 10.1101/gr.2203804 |
| [59] |
|
| [60] |
|
| [61] |
Hidalgo C A, Blumm N, Barabási A L, Christakis N A 2009 PLoS Comput. Biol. 5 e1000353
|
| [62] |
Hannum G, Srivas R, Guénolé A, van Attikum H, Krogan N J, Karp R M, Ideker T 2009 PLoS Genet. 5 e1000782
|
| [63] |
Chuang H Y, Lee E, Liu Y T, Lee D, Ideker T 2007 Molecular Systems Biology 3 140 DOI: 10.1038/msb4100180 |
| [64] |
|
| [65] |
|
| [66] |
Srihari S, Yong C H, Wong L 2017 Computational prediction of protein complexes from protein interaction networks Association for Computing Machinery DOI: 10.1145/3064650 |
| [67] |
|
| [68] |
Pereira Leal J B, Enright A J, Ouzounis C A 2004 PROTEINS: Structure, Function, and Bioinformatics 54 49 DOI: 10.1002/prot.10505 |
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
Getz G, Vendruscolo M, Sachs D, Domany E 2002 Proteins: Structure, Function, and Bioinformatics 46 405 DOI: 10.1002/(ISSN)1097-0134 |
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
Srihari S, Leong H W 2012 International Journal of Bioinformatics Research and Applications 8 286 DOI: 10.1504/IJBRA.2012.048962 |
| [79] |
|
| [80] |
Altaf-Ul-Amin M, Shinbo Y, Mihara K, Kurokawa K, Kanaya S 2006 BMC Bioinformatics 7 207 DOI: 10.1186/1471-2105-7-207 |
| [81] |
|
| [82] |
|
| [83] |
|
| [84] |
Wang H, Kakaradov B, Collins S R, Karotki L, Fiedler D, Shales M, Shokat K M, Walther T C, Krogan N J, Koller D 2009 Mol. Cell Proteomics 8 1361 DOI: 10.1074/mcp.M800490-MCP200 |
| [85] |
|
| [86] |
Radicchi F, Castellano C, Cecconi F, Loreto V, Parisi D 2004 Proc. Natl. Acad. Sci. USA 101 2658 DOI: 10.1073/pnas.0400054101 |
| [87] |
|
| [88] |
Fouss F, Pirotte A, Renders J, Saerens M 2007 IEEE Transactions on Knowledge and Data Engineering 19 355 DOI: 10.1109/TKDE.2007.46 |
| [89] |
|
| [90] |
|
| [91] |
|
| [92] |
|
| [93] |
|
| [94] |
|
| [95] |
|
| [96] |
|
| [97] |
|
| [98] |
Vassura M, Margara L, Di Lena P, Medri F, Fariselli P, Casadio R 2008 IEEEACM Trans. Comput. Biol. Bioinform. 5 357 DOI: 10.1109/tcbb.2008.27 |
| [99] |
|
| [100] |
|
| [101] |
Zhang C, Mortuza S M, He B, Wang Y, Zhang Y 2018 Proteins 86 1 136 DOI: 10.1002/prot.25414 |
| [102] |
Baú D, Martin A J M, Mooney C, Vullo A, Walsh I, Pollastri G 2006 BMC Bioinformatics 7 402 DOI: 10.1186/1471-2105-7-402 |
| [103] |
Kukic P, Mirabello C, Tradigo G, Walsh I, Veltri P, Pollastri G 2014 BMC Bioinformatics 15 6 DOI: 10.1186/1471-2105-15-6 |
| [104] |
Walsh I, Baù D, Martin A J M, Mooney C, Vullo A, Pollastri G 2009 BMC Structural Biology 9 5 DOI: 10.1186/1472-6807-9-5 |
| [105] |
|
| [106] |
Martelli P L, Fariselli P, Malaguti L, Casadio R 2002 Protein Engineering, Design and Selection 15 951 DOI: 10.1093/protein/15.12.951 |
| [107] |
Ceroni A, Passerini A, Vullo A, Frasconi P 2006 Nucleic Acids Res. 34 W177 DOI: 10.1093/nar/gkl266 |
| [108] |
|
| [109] |
|
| [110] |
|
| [111] |
Schaarschmidt J, Monastyrskyy B, Kryshtafovych A, Bonvin A 2018 Proteins 86 1 51 DOI: 10.1002/prot.25407 |
| [112] |
|
| [113] |
|
| [114] |
|
| [115] |
Senior A W, Evans R, Jumper J, Kirkpatrick J, Sifre L, Green T, Qin C, Žídek A, Nelson A W, Bridgland A 2019 Proteins: Structure, Function, and Bioinformatics 87 1141 DOI: 10.1002/prot.v87.12 |
| [116] |
|
| [117] |
|
| [118] |
Zellner H, Staudigel M, Trenner T, Bittkowski M, Wolowski V, Icking C, Merkl R 2012 Proteins 80 154 DOI: 10.1002/prot.23172 |
| [119] |
|
| [120] |
|
| [121] |
|
| [122] |
|
| [123] |
Singh G, Dhole K, Pai P P, Mondal S 2014 SPRINGS: prediction of protein–protein interaction sites using artificial neural networks Report No. 2167–9843 DOI: 10.7287/peerj.preprints.266v2 |
| [124] |
|
| [125] |
|
| [126] |
|
| [127] |
|
| [128] |
|
| [129] |
|
| [130] |
|
| [131] |
Geng H, Lu T, Lin X, Liu Y, Yan F 2015 Biochemistry Research International 2015 978193 DOI: 10.1155/2015/978193 |
| [132] |
|
| [133] |
|
| [134] |
|
| [135] |
|
| [136] |
|
| [137] |
|
| [138] |
|
| [139] |
|
| [140] |
|
| [141] |
|
| [142] |
|
| [143] |
|
| [144] |
|
| [145] |
Krizhevsky A, Sutskever I, Hinton G E Advances in Neural Information Processing Systems 1097 1105 DOI: 10.1186/s40709-016-0046-7 |
| No Suggested Reading articles found! |
|
|
Viewed |
|
|
|
Full text
|
|
|
|
|
Abstract
|
|
|
|
|
Cited |
|
|
|
|
Altmetric
|
|
blogs
Facebook pages
Wikipedia page
Google+ users
|
Online attention
Altmetric calculates a score based on the online attention an article receives. Each coloured thread in the circle represents a different type of online attention. The number in the centre is the Altmetric score. Social media and mainstream news media are the main sources that calculate the score. Reference managers such as Mendeley are also tracked but do not contribute to the score. Older articles often score higher because they have had more time to get noticed. To account for this, Altmetric has included the context data for other articles of a similar age.
View more on Altmetrics
|
|
|