




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Passivation of carbon dimer defects in amorphous SiO2/4H–SiC (0001) interface: A first-principles study
Cite this Article
Zhang Yi-Jie, Yin Zhi-Peng, Su Yan, Wang De-Jun. Passivation of carbon dimer defects in amorphous SiO2/4H–SiC (0001) interface: A first-principles study. Chinese Physics B, 2018, 27(4): 047103 
Permissions
Passivation of carbon dimer defects in amorphous SiO2/4H–SiC (0001) interface: A first-principles study
† Corresponding author. E-mail:
Abstract
An amorphous SiO2/4H–SiC (0001) interface model with carbon dimer defects is established based on density functional theory of the first-principle plane wave pseudopotential method. The structures of carbon dimer defects after passivation by H2 and NO molecules are established, and the interface states before and after passivation are calculated by the Heyd–Scuseria–Ernzerhof (HSE06) hybrid functional scheme. Calculation results indicate that H2 can be adsorbed on the O2–C=C–O2 defect and the carbon–carbon double bond is converted into a single bond. However, H2 cannot be adsorbed on the O2–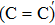
1. Introduction
Semiconductor silicon carbide (SiC) with a wide band gap has the characteristics of high thermal conductivity, high carrier saturation velocity, and large critical breakdown field strength, which make it a prominent candidate for replacing the traditional Si material.[1–3] As a power control device, the SiC metal–oxide–semiconductor field-effect transistor (MOSFET) is one of the research focuses among the several SiC devices thus far.[4–6] However, the density of states (DOS) in the SiC metal–oxide–semiconductor (MOS) device fabricated through a conventional thermal oxidation process is extremely high; this phenomenon greatly reduces the channel mobility by capturing and scattering carriers in the channel, thereby deteriorating the performance of SiC MOS devices seriously.[7–13] The origin of the density of interface traps (DIT) can be attributed to the dangling bonds, carbon clusters, and near-interface oxide traps at and near the SiO2/SiC interface.[14–18] Carbon cluster is a critical defect contributing to the density of interface states. Pensl et al.[19] reported that the minimum DOS at the SiO2/SiC interface is at least one order of magnitude higher than that at the Si/SiO2 interface and concluded that the excess DOS is caused by the residual carbon–carbon double bonds (C cluster or graphite-like structure). Afanas’ev et al.[20] determined that the carbon aggregation at the SiO2/SiC interface is one of the two major factors leading to the high density of interface states. At present, the method to reduce DIT is to eliminate the SiO2/SiC interface defects by the passivation process. Therefore, exploring the mechanism for passivating SiO2/SiC interface defects is important.
Considerable researches have been conducted on the passivation process of the SiO2/SiC interface. Fukuda et al.[21] studied the high-temperature annealing process in H2 atmosphere of SiC MOSFET, which can reduce the DIT in the SiO2/SiC interface. Cantin et al.[22] also used a high-temperature annealing process in H2 to passivate the carbon dangling bonds of the SiO2/SiC interface; however, they found that the H atom presents a depassivation process at high temperature, thereby reducing the passivation effect. Although H2 passivation process is still under debate,[23–25] NO passivation is considered an effective passivation method to reduce DIT. Chung et al.[26] used the NO annealing process to passivate the SiO2/SiC interface, and the results showed that NO can reduce the DIT at the SiO2/SiC interface. Then they proposed that the NO passivation SiC/SiO2 interface is achieved by bonding some N atoms with C atoms. In recent years, the theoretical research on SiO2/SiC passivation has gradually been a focus due to the decrease in calculation cost and improvement in calculation condition. Knaup et al.[27] studied the mechanism for dry oxidation by calculation and identified some defect configurations relating to carbon atoms (interstitial carbon atom and carbon dimers), which can be produced during SiC thermal oxidation. Devynck et al.[28] calculated an abrupt interface structure at the SiO2/SiC interface by first principle. Li et al.[29] constructed various defect structures at the SiO2/SiC amorphous interface based on the amorphous interface structure and combined their study with Knaup’s. The energy band and DOS were analyzed, and the authors suggested that the carbon dimers and the silicon vacancy among the defects are probably the main reasons for introducing DOS into the SiO2/SiC interface.
From the considerable researches on the defects and passivation process of the SiO2/SiC interface in experiments, few theoretical calculations are available with respect to the passivation structure of the SiO2/SiC interface. Among the many defects at the SiO2/SiC interface, the carbon dimer defect is a typical type of carbon cluster defect with a carbon–carbon double bond, and some studies have shown that it is an important defect contributing to the DOS into the band gap of SiC.[19,30,31] In this study, we establish an amorphous SiO2/4H–SiC (0001) interface model with carbon dimer defects based on the research of SiO2/SiC interface defects by Li et al.[29] The structures of the carbon dimer defects that passivated through the H2 and NO molecules are calculated by standard density functional theory (DFT). The HSE06 hybrid functional method is then used to calculate the DOS and the partial charge density. The passivation mechanism is elaborated by comparing the DOS and the partial charge density before and after passivating through H2 and NO molecules.
2. Calculation methods
In this expression,
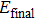
and
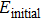
are the total energy before and after passivation, respectively. The integer ni represents the number of added atoms of type i when creating a structural passivation, and the
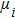
is the chemical potential used for i (H, N, or O).
In this work, the Vienna ab initio simulation package software based on DFT was used for the simulation.[32,33] For the calculation of structural relaxation, the electron exchange correlations were obtained using the generalized gradient approximation of the PBE function.[34] The cutoff energy for the wave function of the valence electron was set to be 400 eV. The Monkhorst–Pack k-point mesh was sampled with a constant separation of 0.04 Å−1 in the Brillouin zone.[35] All structural models were fully relaxed using standard DFT until the atomic force was less than 0.005 eV/Å. Furthermore, the stability of complete passivation structure has been checked by the ab initio molecular dynamics. It can ensure the stability in the high temperature annealing process.
The formation energy[36–38] of structure after passivation is computed from
 |
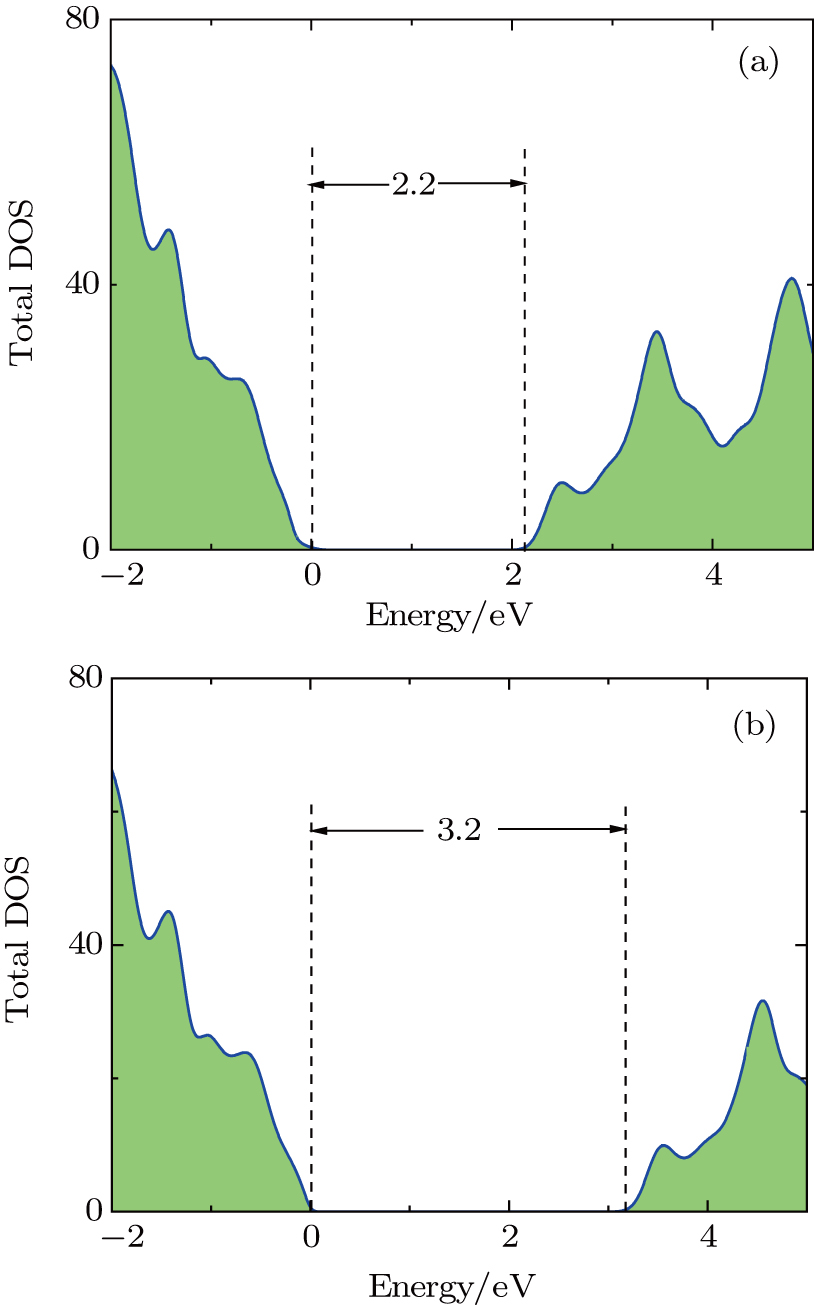
The standard DFT and the HSE06 hybrid functional scheme were employed to calculate the DOS of the 4H–SiC band gap. The results showed that the standard DFT underestimated the band gap of 4H–SiC as shown in Fig.
 | Fig. 1. (color online) DOSs of 4H–SiC calculated (a) by standard DFT, in which the band gap is 2.2 eV, and (b) by HSE06 hybrid function, in which the band gap is 3.2 eV. The 4H–SiC band gap obtained in the experiment is 3.26 eV. Level of 0 eV is marked as valence band energy of SiC. |
3. Results and discussion
3.1. Defect structures of carbon dimer and DOS
According to the study of Li et al.,[29] we establish the amorphous SiO2/4H–SiC interface structure as shown in Fig.
 | Fig. 2. (color online) (a) Interface structure of amorphous SiO2/4H–SiC. (b) Interface structure of SiO2/SiC containing a carbon dimer defect, named the O2–C = C–O2 defect. (c) Another interface structure of SiO2/SiC containing a carbon dimer defect, named the O2–(C = C)–O2 defect. |
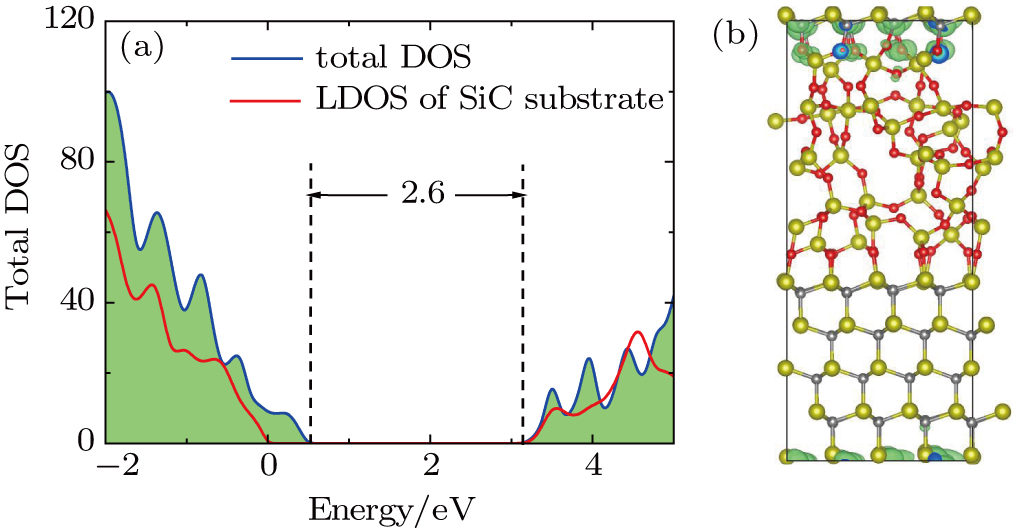
After we obtain the structural model, we calculate the DOSs for these three structures by using the HSE06 hybrid function. In Fig.
 | Fig. 3. (color online) (a) DOS in amorphous SiO2/4H–SiC interface. Maximum of valence band of the SiC substrate is set to be 0 eV. The red line represents partial density of states (PDOS) of SiC substrate, and the blue line denotes the three total DOSs of the interface structure. (b) Partial charge density diagram of energy band in amorphous SiO2/4H–SiC interface in energy level range of 0 eV–0.6 eV. Isosurface value is set to be 0.03 e/Bohr3. |
Figure
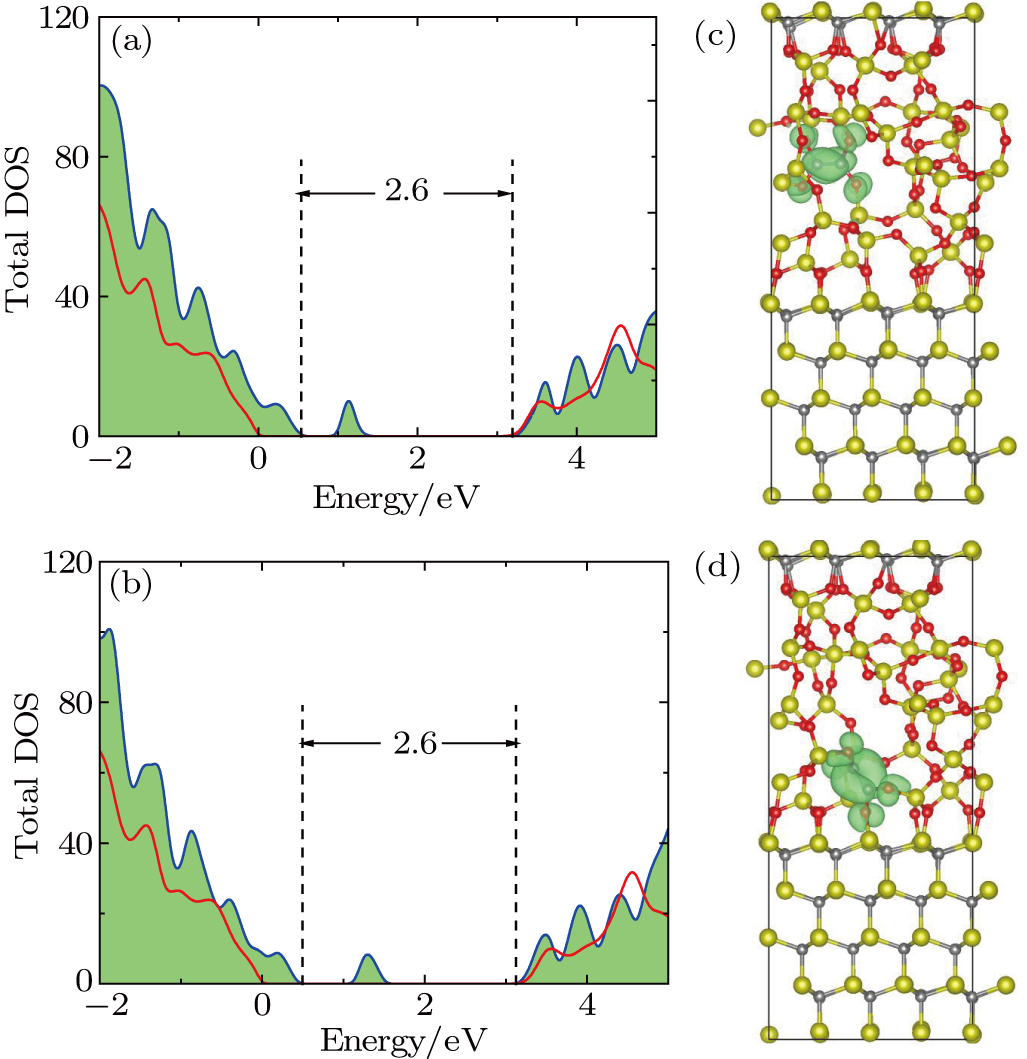 | Fig. 4. (color online) DOSs of (a) O2–C = C–O2 defect and (b) O2–
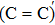
|
3.2. Defect structure and DOS of carbon dimer defects after H2 passivation
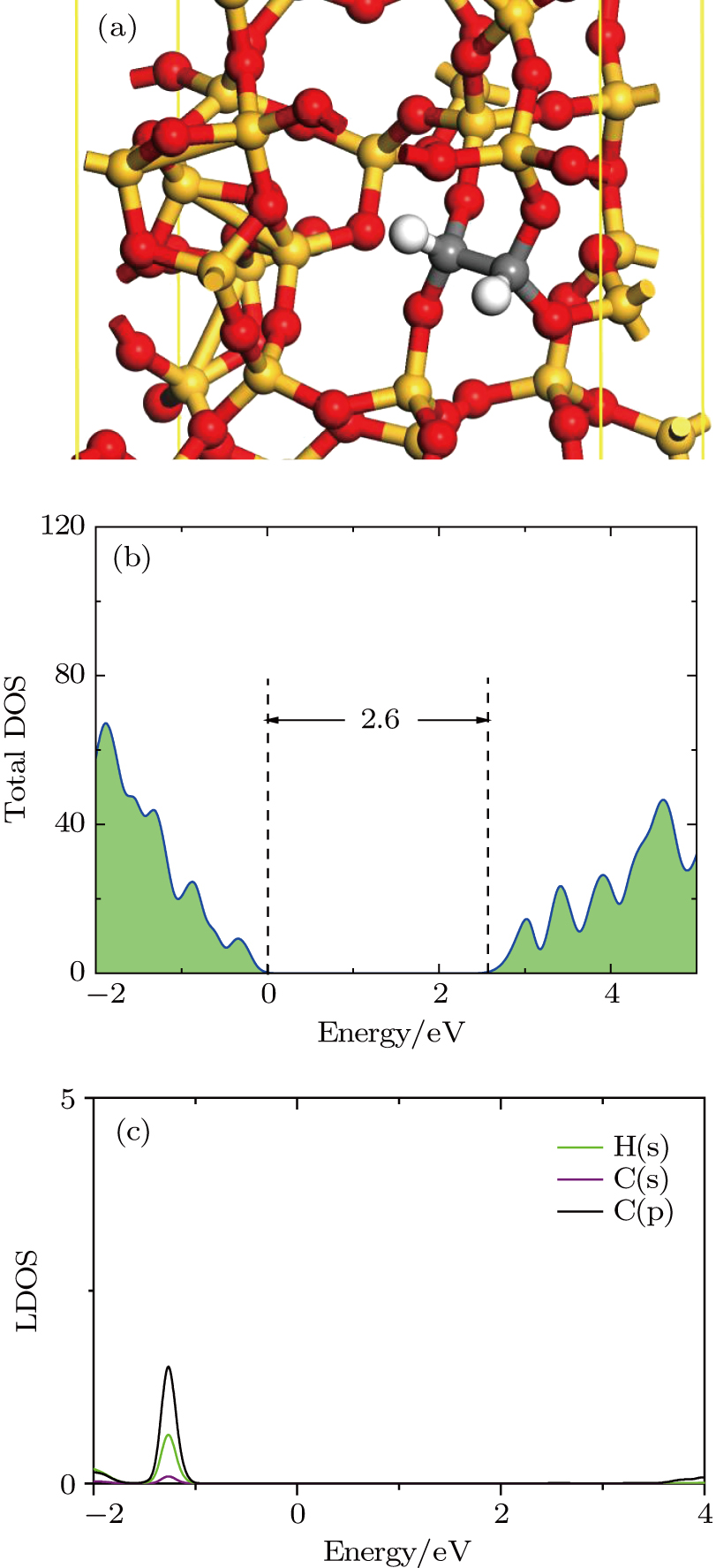
We study the carbon dimer defect structures passivated by H2. After structure optimization, we obtain the structure and its DOS as shown in Fig. 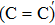
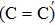
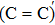

 | Fig. 5. (color online) (a) O2–C = C–O2 defect structure after H2 passivation, (b) its total DOS, and (c) PDOSs of atoms in the passivation region. |
3.3. Defect structure and DOS of carbon dimer defects after NO passivation
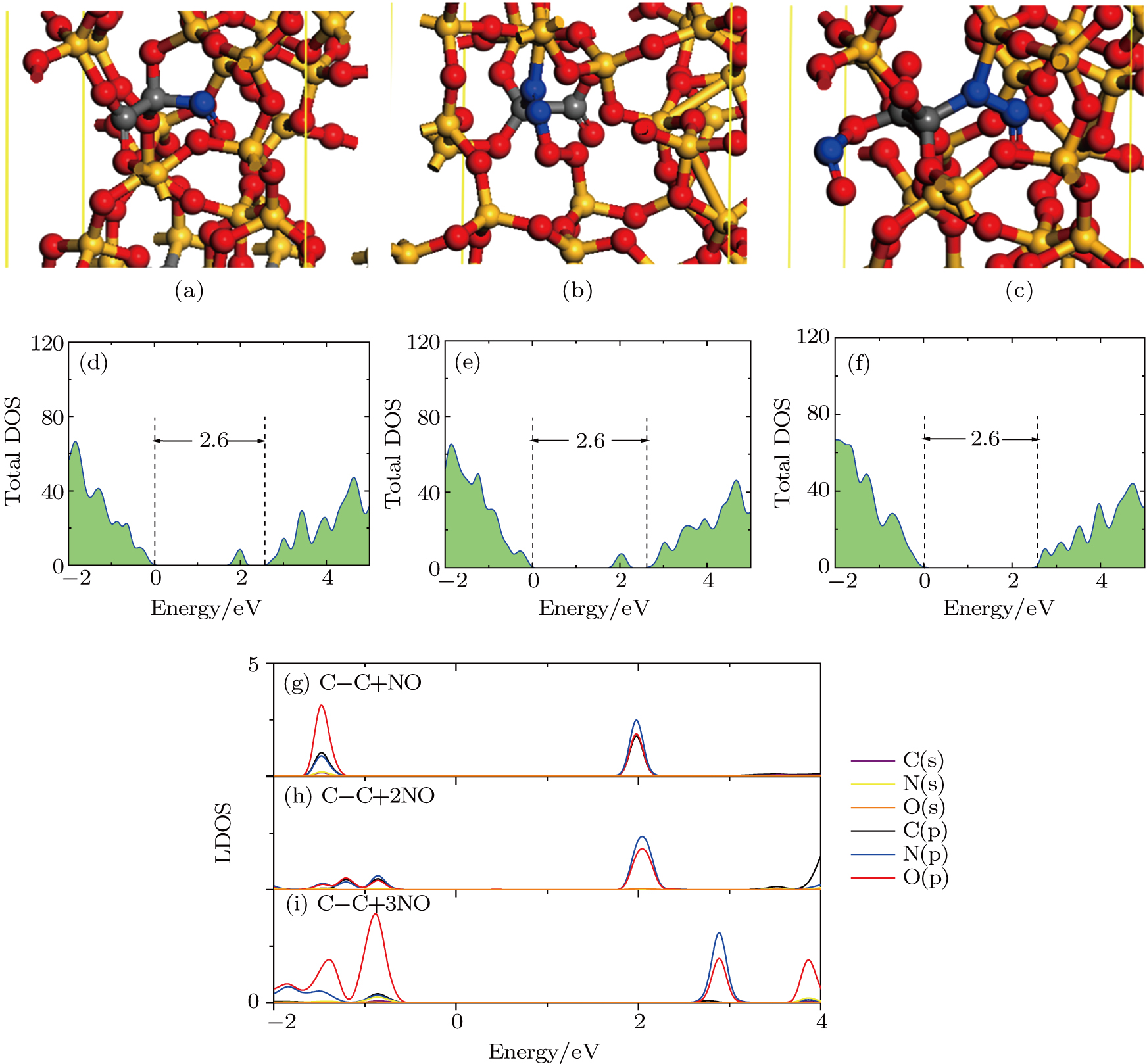
The results of O2–C = C–O2 defect after NO passivation are shown in Fig.
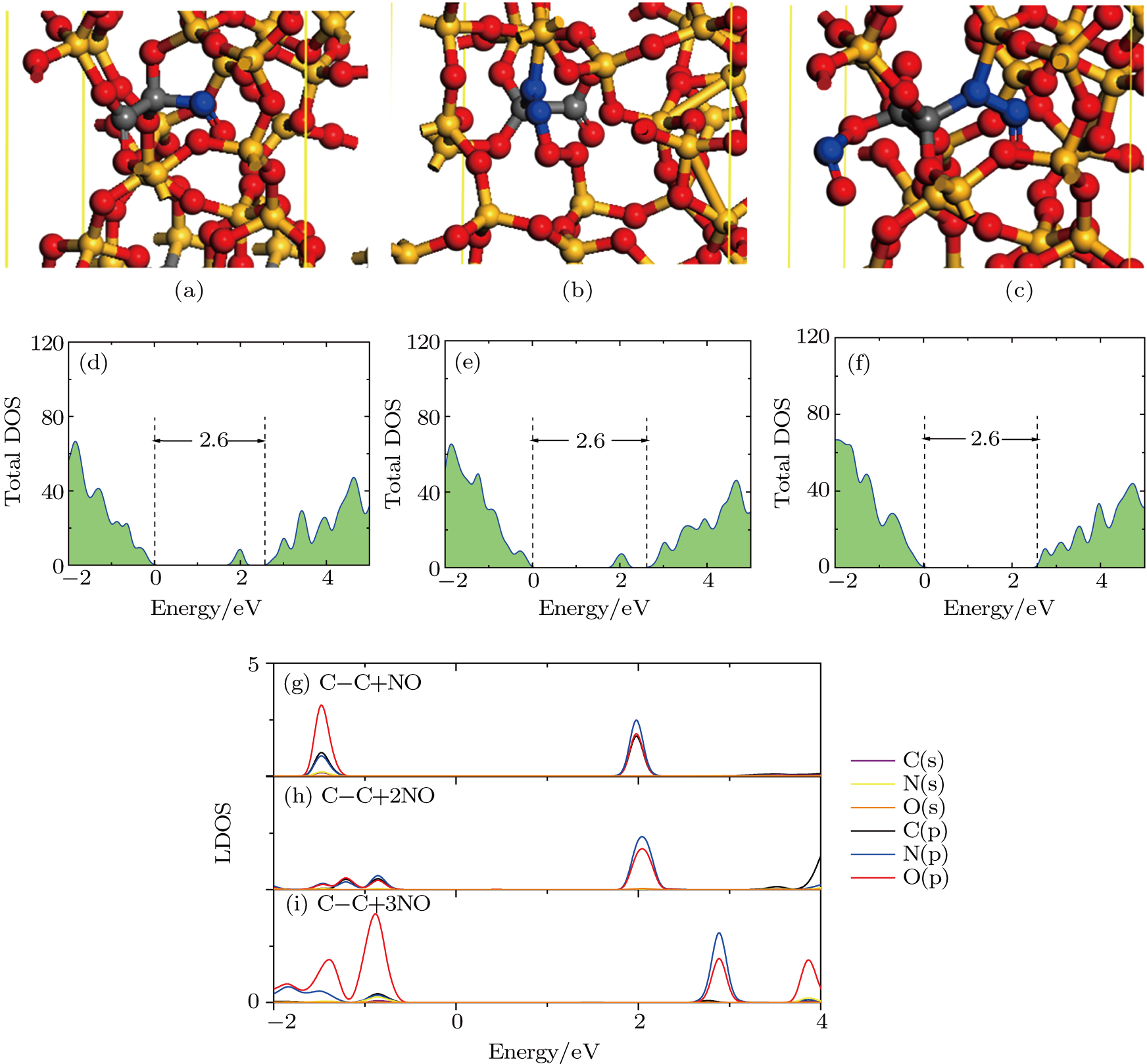 | Fig. 6. (color online) Structure diagrams and DOSs of the O2–C = C–O2 defect after NO passivation, showing passivation structures of (a) C–C + NO, (b) C–C + 2NO, and (c) C–C + 3NO, total DOSs of (d) C–C + NO, (e) C–C + 2NO, and (f) C–C + 3NO passivation structures, PDOSs of atoms in the passivation region of (g) C–C + NO, (h) C–C + 2NO, and (i) C–C + 3NO passivation structures. |
The results of the O2–(C = C)’–O2 defect after NO passivation are shown in Fig. 

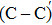
 | Fig. 7. (color online) Structure diagrams and DOSs of the O2–






|
For further calculations, we obtain another type of NO passivation O2–
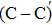
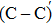
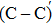
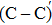
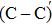


 | Fig. 8. (color online) Structure diagrams and DOSs of the O2–


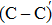
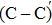
|
The process of passivating carbon dimer defects at the SiO2/SiC interface by NO is more complicated than by H2. The key of passivating carbon dimer defects is to transform or break the carbon–carbon double bond. Moreover, the defects should be completely passivated; otherwise, some intermediate structures would also introduce DOS into the band gap.
3.4. Partial charge density of incompletely passivated interface
We calculated the partial charge densities of the four intermediate structures with incomplete passivation, namely, C–C + NO, C–C + 2NO, 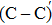
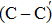


 | Fig. 9. (color online) Partial charge density diagrams of the intermediate structure in the band gap of SiO2/4H–SiC after incomplete passivation by NO, showing intermediate structures of (a) C–C + NO, (b) C–C + 2NO, (c) 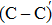

|
4. Conclusions
The defect structure and interface states, as well as partial charge density, in the SiO2/4H–SiC (0001) interface have been calculated and compared by using the first principle. The results show that H2 can be adsorbed on the O2–C = C–O2 defect to form a passivation structure but it has no effect on the O2–
Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] | |
| [33] | |
| [34] | |
| [35] | |
| [36] | |
| [37] | |
| [38] | |
| [39] | |
| [40] | |
| [41] |