1. IntroductionTo reveal the structural features of materials at multiple scales, various characterization techniques have been employed, including x-ray diffraction (XRD), neutron powder diffraction (NPD), x-ray absorption spectroscopy (XAS), and electron microscopy. XRD and NPD have been implemented in structural characterization to obtain the crystallographic information, such as structural features and lattice parameters. XAS has been widely applied to probe the evolution of the chemical environment and electronic structure. However, these techniques suffer from a few limitations due to their lower spatial resolutions.
Electron microscopy is a broad term that includes scanning electron microscopy (SEM), scanning tunneling microscopy (STM), transmission electron microscopy (TEM), and scanning transmission electron microscopy (STEM). SEM is mainly utilized for surface morphological characterization and STM is used to reveal the surface atomic and electronic structure. In particular, TEM and STEM have superior abilities in detecting the structure of materials with higher spatial resolution. In addition, they could provide information related to chemical composition and electronic structure. The aberration corrector, originally introduced by Scherzer,[1] makes it possible to achieve sub-angstrom resolution without using complex computation.[2–6] Given these prominent advantages, TEM and STEM have been used in many fields, such as physics, chemistry, material science, etc.[7,8] This review mainly focuses on HAADF and ABF imaging techniques and their applications in characterization of electrode materials for Li-ion batteries.
2. Brief history of TEM and STEMTransmission electron microscopy was developed between the 1920s and 1930s. The French physicist De Broglie discovered the wave nature of electrons in 1924, proved by electron diffraction. The German scientist Busch suggested that an axisymmetrical magnetic field could focus the electron beam, providing the theoretical basis of TEM. Afterward, Knoll and Ruska used these concepts to successfully build the first TEM in 1934 Meanwhile, the theory of high-resolution transmission electron microscopy (HRTEM) was established and improved, promoting the field application of HRTEM. The atomic column of most crystals can be imaged by HRTEM. However, the intrinsic aberration of the lens, especially the spherical aberration, limits the resolution. The development of aberration corrector paved a great step forward for enhancing resolution.
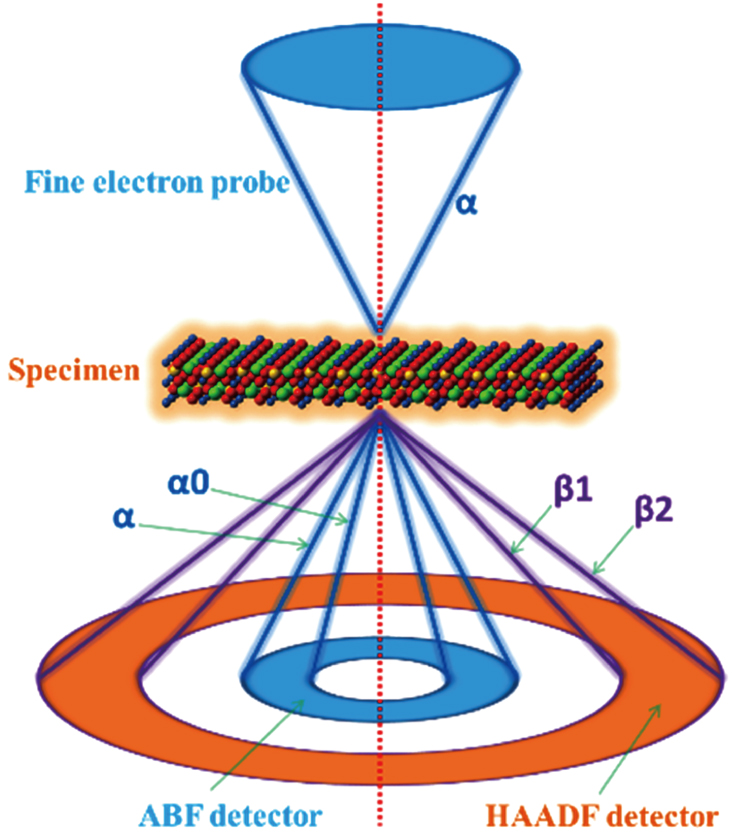
Compared with TEM, a scanning focused incident electron beam rather than a static one is used in STEM. Moreover, an annular detector centering bright field is employed to collect scattered electrons in STEM. Pennycook et al. proposed high angle annular dark field (HAADF) imaging technique for STEM to obtain the atomic number (Z) contrast[9] and the direct visual interpretability of images and insensitivity to sample thickness were significantly advanced. The signal intensity of the atomic columns of HAADF was found almost proportional to Z1.7.[9,10] However, when the light and heavy elements are present simultaneously, the weak signals from light elements are completely overwhelmed by the strong contrast issued from heavy elements.[10] Light elements have been shown to play important roles in advanced functional materials and devices. For example, the migration of Li atoms was found crucial in mechanisms of Li-ion batteries. Therefore, a great challenge is to make light and heavy elements visible simultaneously. This difficulty was solved in 2009 when Okunishi et al. reported an innovative imaging technique, called annular bright-field (ABF), where light and heavy elements in iron oxide, silicon nitride, and strontium titanate were simultaneously visualized.[11] After the first successful demonstration, increasing attention was paid to ABF imaging. Numerous experiments and theoretical studies confirmed that this imaging mode could visualize light and heavy elements simultaneously within a relative large range of specimen thickness and defocus value.[12–17] Using ABF imaging, numerous unknown structures have been discovered one after another. Nowadays, the advanced imaging techniques are utilized in various fields.
4. Applications in structural characterization of lithium ion battery cathode materialsLithium-ion batteries (LIBs) were first commercialized in 1991 by Sony and currently used in many fields, such as portable electronics, electric vehicles, and smart grids. This is due to their high energy and power densities, as well as their superior cyclic stabilities and safety performances.[27–29] The cathode materials are vital components in the construction of LIBs, largely affecting the battery performances. The conventional cathode materials include olivine compounds LiMPO4 (M = Fe, Mn, Ni, Co, etc.), layered compounds LiMO2 (M = Co, Ni, Mn, etc.), and spinel compounds LiM2O4 (M = Mn, etc.). To improve the properties of LIBs and meet demands for emerging markets, understanding the structure and mechanism of the electrode materials is necessary. Although the atomic behavior of the Li ion primarily determines the electrochemical performances of LIBs, such information is hardly obtained by TEM technique due to the weak electron scattering of light atoms.[30] Considering the superior performances of ABF imaging in terms of simultaneous visibility of light and heavy atoms, ABF has been employed to reveal structural features of electrode materials and electrochemical reaction mechanism in LIBs.[31,32] In addition, the HAADF imaging technique has been widely used for investigating the structural evolution of electrode materials.
4.1. Olivine compound LiFePO4Since LiFePO4 was first reported as a cathode material for LIBs,[33] it has increasingly attracted attention due to its low cost, safety performance and cyclic stability. Although the practical applications of LiFePO4 in cathodes of LIBs have been realized for many years, several problems remained unsolved. For instance, the intrinsic low electronic conductivity[34–36] and poor ionic transport properties[37–40] were considered significant limitations in achieving high rate performances. Nanostructure[41–44] and surface coating[45–48] could overcome these drawbacks to some extent. The microscopic mechanism of structural evolution became essential in revealing the electrochemical reaction mechanisms and improving the properties of LIBs. An obvious voltage plateau appeared during the charge and discharge processes of LIBs based on LiFePO4 cathodes. This suggested that a two-phase reaction in the LiFePO4 cathode took place. Many models have been developed to explain the two-phase reaction mechanism, including the core–shell model,[33] the shrinking model,[49] the mosaic model,[50] the new core–shell model,[51] and the domino-cascade model.[52] But all of these models failed to provide details on the structure of resulting two-phase interface region, essential to explain the two-phase reaction mechanism.
The lithium staging structure in partially delithiated LixFePO4 nanowires (x ≈ 0.5, d = 65 nm) was observed for the first time by an aberration-corrected ABF-STEM (Fig. 5).[53] Compared to pristine LiFePO4 (Fig. 5(a)), the Li-extraction sites in half-charged LiFePO4 are marked by orange circles (Fig. 5(c)). The lithium staging structure looks obvious, and the results are different from the previous models. Later, a single-phase transformation path was demonstrated to exist at very low overpotential during lithiated and dislithiated processes to yield a metastable staging-like atomic configuration in canonical Monte Caro simulations.[54] Meanwhile, the discovery of the metastable phase provided explanations regarding the high rate capability of LiFePO4. In addition, the formation mechanism of the lithium staging structure and the factors influencing its formation were investigated. The atomic structure of partially chemically dislithiated 2% Nb-doped LiFePO4 with particle size of ∼ 200 nm was revealed by ABF imaging and the lithium staging structure was detected.[55] These studies showed that doping of Nb did not affect the formation of the lithium staging structure, indicating that the staging single phase was an intermediate or intrinsic metastable phase.
On the other hand, the lithium staging structure was used as “buffer” to relax volume expansion (about 6.8%) and facilitate the migration of Li ions. This supported the fact that the formation of the lithium staging structure is advantageous to the electrode kinetics. The phenomenon of staging LiFePO4 with different sizes (70 nm and 50 nm) was detected by ABF imaging.[56] The ABF-STEM images of partially dislithiated 70 nm LiFePO4 are shown in Fig. 6. The lithium staging structure mainly existed in the region between LiFePO4 and FePO4, creating coexisting LiFePO4/staging structure/FePO4. Two LiFePO4 samples with different sizes demonstrated the effect of size on the lithium staging region. In other words, the zone of the lithium staging structure becomes narrower as the LiFePO4 size increases.
The theoretical calculation regarding the formation of such staging structures was performed by density functional theory,[57] showing that the staging structure in partially delithiated LiFePO4 is thermodynamically metastable but in the kinetically controlled state. A dual-interface model was also proposed and the results were consistent with experimentally observed three-phase coexisting LiFePO4/staging structure/FePO4. The formation of the lithium staging structure was mainly ascribed to the Fe center mediating the interlayer Li–Li interaction, originating from the localized nature of Fe 3d electrons. The effective oxidation state of Fe redox depended on the configuration of the Li ions. Conversely, the diffusion of Li ions was influenced by the oxidation state of Fe but the size effect was not taken into consideration in this work. Overall, ABF imaging of STEM coupled with other characterization techniques would reveal the microscopic mechanism of structural evolution in LiFePO4 in a step-by-step process.
4.2. Layered compounds LiCoO2 and Li2MnO3LiCoO2 was first reported by Mizushima as a cathode material with high volumetric energy density.[58] LiCoO2 cathode was employed in the first generation of commercial LIBs and still dominates the portable devices market. However, no more than 0.5 mole of Li ions can be extracted per mole of Co to maintain structural stability during electrochemical cycling, causing extremely low potential capacities of LIBs. Three kinds of layered structures of LiCoO2 are O1-type, O2-type, and O3-type. The stacking sequences of O1, O2, and O3 are ABAB, ABAC, and ABCABC, respectively.[58–60] It has been observed that O2-LixCoO2 can be formed in electrochemically cycled O3-LiCoO2 electrodes via HAADF and ABF-STEM.[61] When LiCoO2 cathodes are charged to 4.5 V (x = 0.5), the structure of LiCoO2 should undergo a transformation from O3-type to O1-type (Fig. 7). In Figs. 7(b) and 7(c), the contrast of Co columns (pink line: ○) is obviously different from that of the black line (☆) acquired by the pristine LiCoO2 ABF micrograph. The Co atoms are marked with Co1 and Co2, denoting different chemical environments of Co ion after delithiation at high voltage.
The different contrast of the Co atom is attributed to the interaction with the nearest Li ion and vacancy, providing information about the electron distribution and bond length distortion after Li ion extraction. In Fig. 7(d), the Li ion columns (Li1 and Li2) in the blue line (○) are also distinct from those in the black line (☆) from the pristine material, whose configuration is similar to the arrangement of the Li ion and vacancy in Fig. 7(a). Such configuration was observed along the (101) plane in the O1-LiCoO2 lattice. As shown in Fig. 8, the phase transition that occurred during the first cycle was visible by HAADF-STEM. The phase transition from O3-LiCoO2 to O1-LiCoO2 was observed at x = 0.5 in nanosized LixCoO2. This result does not agree well with the known phase diagram of LixCoO2, where the O3 phase was kept before x = 0.5 and the initial formation of O1 occurred at x = 0.3.[62–64] Afterwards, the O2 phase started to form when the charge state was discharged to 3.0 V. The O2-type is metastable generally created by ionic exchange from NaCoO2[59,60] and O3-type is a stable state. Earlier, it was thought that the phase transition processes of O2 and O3 were separate, meaning that they could not convert into each other once synthesized. This finding connected the two separated LiCoO2 systems to each other, at least for nanoscale particles (50 nm) used experimentally.
All-solid-state batteries equipped with solid-state electrolytes are effective approaches to address safety problems related to flammable organic liquid electrolytes. Solid-state electrolytes make it possible for lithium to be used in anodes and high voltage cathodes materials at the same time. The structure evolution of LiCoO2 in all-solid-state LIB and structure at high voltage conditions have been experimentally studied in situ by STEM imaging.[65] The working all-solid-state LIB shown in Fig. 9(a) was designed, and electrochemical delithiation was directly observed at the atomic scale by employing a state-of-the-art chip by aberration-corrected STEM for the first time. The all-solid-state LIBs with gold anodes, LiCoO2 cathodes, and Y and Ta-doped LLZO (Li6.75La2.84Y0.16Zr1.75Ta0.25O12) as the solid-state electrolyte (SSE) were built on micro-electro-mechanical system (MEMS) device nanochips using FIB milling.[66–68]
ABF and HAADF images of pristine LiCoO2 cathodes are shown in Figs. 9(c) and 9(d), demonstrating the layered structures of LiCoO2 cathodes. As shown in Fig. 10, after high voltage delithiation, the pristine single crystal LiCoO2 became a nanosized polycrystal connected by coherent twin boundaries and antiphase domain boundaries. Figures 10(b)–10(e) also reveal that only two kinds of crystal orientations in the delithiated LiCoO2 cathode existed and the original 109.5° coherent twin boundary in the pristine LiCoO2 cathode increased to 112° due to the extraction of lithium ions. Contrasts in lithium layer in both HAADF and ABF micrographs are visible for both boundaries, suggesting that heavy atoms were present in the lithium layer. In this case, they concerned cobalt ions due to a phase transition in the layer structure, spinel and rock salt. Meanwhile, the layer spacing increased by 2.7% (±0.4%) due to the extraction of lithium ions and probably accumulated lithium or cobalt ions at the domain boundaries.[69–72]
To elucidate the formation of both boundaries, three models with different lithium concentrations were utilized to calculate the formation energies. The interface energies of coherent twin boundaries and antiphase domain boundaries with different Li concentrations suggested that the antiphase domain boundary is less likely to form than the coherent twin boundary during the delithiation process, which is consistent with the HAADF micrographs. Thus, this work not only boosted the development of all-solid-state batteries but also introduced the state-of-the-art chip to study electrochemical processes. Furthermore, the results of the electrochemical measurements will be improved due to the decreased contact resistances.
Apart from LiCoO2, a kind of compounded material composed of Li2MnO3 and LiMO2 (M = Mn, Ni, Co, Fe, Cr, etc.) layers were reported and identified as xLiMO2 · (1 − x)Li2MnO3 or Li-rich layered materials. LIBs with these cathode materials can provide much higher energy densities than those with LiCoO2.[73,74] Li2MnO3 is considered as the end member of the xLiMO2 · (1 − x)Li2MnO3 compound family, extensively investigated for applications as cathodes in LIBs.[74–76] The structure of Li2MnO3 is a layered Li[Li1/3Mn2/3]O2 structure, where Li+ and Mn4+ ions occupy the octahedral interstices of the cubic closely-packed oxygen lattice. Because the content of Li+ is rich, they are thus called Li-rich layered materials. However, delithiation is difficult in Li2MnO3, leading to much lower experimental capacities than the theoretical values.[77,78] Some studies reported the observation of crystal structure transformation of Li2MnO3 implemented by the aberration-corrected STEM and high-resolution XRD, revealing the coexistence of monoclinic spinel and rock salt structures.[79] Hence, localized and inhomogeneous structural evolution was discovered.
The transition metal atoms occupying the Li plane is obvious, and considered as a key step in generating the rock salt or spinel structure. Many small regions marked by strain are surrounded by darker contrast. Meanwhile, the Li2MnO3 regions are adjacent to the transferred regions, pointing to the inhomogeneous nature of structure evolution. High-resolution XRD showed that electrochemical reaction occurred in most particles because Li2MnO3 regions and transferred regions existed in most particles of surface and away from the surface towards the bulk. The result is different from previous conclusions that the electrochemical reactions were confined at the surface in similar layered oxide materials.[80–82] EELS analysis provided solid information regarding valence of elements and their chemical environments. In conclusion, the following transformation sequence was proposed: Li2MnO3 → LixMn4/3O4 → LixMn2O4 → Mn3O4 → MnO. Oxygen trapping appeared inside the particles, producing Mn-deficient topotaxial LixMn2O4 regions. However, O diffusion at the surface of the particles was easier, leading to the formation of MnO. Finally, the observed capacity fading may be attributed to a phase transition.
Inferior rate capacity and voltage decay of Li-rich layered materials during electrochemical cycle are great obstacles in practical applications.[74] These issues are attributed to poor electrode kinetics, where kinetics of Li-rich layered material could even be worse with largely dense particles. Because the Li2MnO3 phase is partially linked to electrochemical activation, several cycles are essential to achieving full activation. Such an electrochemical cycle is harmful to reversible capacity.[83,84] Therefore, tremendous efforts have been made to solve these issues, including by surface coating with Al2O3, AlPO4, AlF3[85,86] and nanosizing,[87,88] which can improve the behaviors of the cathode to some degree.
Some studies demonstrated that the electrochemical activity could be facilitated by structural defects.[84,89,90] Gradient surface Na+ doping achieved by calcination process of Li-rich layered material in a molten NaCl state was thus introduced to enhance the kinetics of Li-rich layered materials.[91] The structure with gradient surface Na+ is shown schematically in Fig. 11. The HAADF image corresponding to the ABF view and ABF profile of Li-rich layered material with gradient surface Na+ doping is presented in Fig. 12. It can be seen from Fig. 12(a) that there is no cation mixture of Li ions and transition metal (TM) ions, because there is no visible contrast in the Li slab. The contrast in the Li slab of ABF induced by TM ions can be excluded. The atomic number of Na was larger than that of Li. The visible contrast in the Li slab of ABF was generated by an increscent averaged atom number. The higher peak of the ABF line profile also indicated the existence of Na+ in the Li slab. All these data observed using aberration-corrected STEM confirmed that doped Na+ occupied the Li sites.
The pinning effects provoked by gradient Na+ doping can stabilize the layered structure by abounding the structural defects. Moreover, this could enlarge the layer-spacing of (003) and (104), as confirmed by XRD. The latter might speed up the diffusion of Li ions in the layered structure. The electrochemical characterizations suggested that when compared with pristine Li2MnO3, the reversible capacity and cyclic stability of Li2MnO3 cathode with gradient surface Na+ doping were highly improved. Doping foreign ions on the surface of electrode materials is, thus, a feasible way to improve electrode kinetics and stabilize the surficial structure. To further advance batteries, the determination of electrochemical mechanism would be crucial to optimize electrode performances.
4.3. Spinel compound LiMn2O4Spinel LiMn2O4 and its derivatives are excellent cathode materials for LIBs due to their high energy densities, low cost, and favorable safety.[92–96] However, the main problem with LiMn2O4 is the serious capacity fading during electrochemical cycling or lengthy storage time, especially at elevated temperatures.[97–100] To overcome these issues, several theories were put forward to explain the phenomena, including electrochemical reaction with electrolytes at high voltage,[101] instability of the two-phase structure at charge state,[102,103] phase transformation from cubic spinel to tetragonal rock-salt structure due to nonequilibrium lithiation,[104] and manganese dissolution.[102,105,106] Even though the structure of LiMn2O4 is relatively stable, migration of Mn ions from octahedral sites to lithium tetrahedral sites in few nanometers surface region during cycling could generate LiMn3O4-like defect-spinel structures.[107,108] The structure distortion at the surface is related to manganese dissolution. Thus, the precise structural evolution in the surface region would be vital in exploring the cause of capacity fading and measures to improve the cycling performance of LiMn2O4 cathodes.
It was reported that an unusual spinel-to-layered transformation of the LiMn2O4 cathode was observed by aberration-corrector STEM, and possible transformation processes were given.[109] Moreover, an atomic-level surface LiMn2O4 cathode subjected to heat-treatment in various atmospheres showed similar transformations.[109] A LiMn2O4 cathode immersed in half-cell was cycled between 3 V and 4.5 V (normal voltage) for six cycles to stabilize, then cycled between 3 V and 4.9 V (high voltage) for ten cycles. The subsequent cycles were implemented between 3 V and 4.5 V. After 100 cycles, no newly formed phases were observed by XRD, and the lattice parameters of LiMn2O4 cathode became smaller than those of pristine materials. This was partially induced by the loss of Mn ions. The XRD results revealed that the spinel structure of LiMn2O4 cathode was relatively stable. Hence, the degradation of cycling performance was probably connected to the local structure of the surface.
The HADDF images of sub-surface and surface of LiMn2O4 cathode cycled in normal voltage window are shown in Fig. 13. The subsurface still maintained the spinel structure, which can be seen from Figs. 13(a) and 13(b) and confirmed by line profile of Figs. 13(e1) and 13(f1). The migration of Mn ions from Mn octahedral sites to lithium tetrahedral sites induced surface distortion, forming defect LiMn3O4-like structures. Compared with ideal LiMn3O4 stimulated structure and line profiles, it was demonstrated that the surface structure is intermediate stage of structure evolution from spinel LiMn2O4 to defect spinel LiMn3O4. As illustrated in Fig. 14, the structure of the bulk kept the standard spinel, and both the distorted surface and subsurface regions after high voltage cycling were larger than those obtained after normal voltage cycling. An unusual layered structure in the surface region appears in Fig. 14(c). The spacing between layers was estimated to be about 4.7 Å, which is comparable to that of many-layered cathode materials.[61,110] During high voltage cycling, Mn ions of the surface LiMn3O4-like structure region from the Mn tetrahedral sites and Mn octahedral sites in some regions fully occupy Mn octahedral sites in other regions nearby, forming a Mn-deficient LiMn3O4-like structure and fresh layered-like structure.
The surface electronic structures of the LiMn2O4 cathode at different electrochemical states were studied by x-ray photoelectron spectroscopy. The existence of Mn3+ on the surface region indicated that oxygen loss may occur on the surface of the LiMn2O4 cathode to reduce Mn4+ ions into Mn3+ compensating charges. During this process, more oxygen may be lost at high cycling voltages. The unusual structure transformation caused by Mn ion migration was related to the reduction of Mn4+ and oxygen loss, confirmed by heat-treatment experiments of the LiMn2O4 cathode. Mn3+ played a key role in the kinetics of the unusual structure transformation in spinel LiMn2O4 cathodes, and the particular atmosphere could control the surface oxygen loss so that the structural transformation could clearly be reconstructed. The unusual spinel-to-layered transformation on the surface of LiMn2O4 cathodes may be a major factor accounting for the degradation of the cycling performance. Some measures can be taken to stabilize the surficial structure to enhance the properties of spinel LiMn2O4 cathodes.
5. PerspectivesThis review is mainly focused on applications of HAADF and ABF of STEM in the characterization of cathode materials of LIBs. For decades, the research and development dealing with LIBs have been focused on rechargeable features. However, little attention was devoted to detecting the mechanisms involved in the redox processes, mainly due to difficulties in locating the Li ions. Li ions play crucial roles in energy storage and transfer in LIBs. The advantage of ABF imaging is visualizing the Li ions, thereby promoting the development of LIBs. Using ABF imaging, many electrochemical reaction mechanisms were revealed and design guidance was outlined to improve the properties of LIBs to meet the increasing demands of clean energy and environmental protection. All-solid-state batteries attracted more attention in terms of high energy density and outstanding safety. However, many problems emerged due to the use of solid electrolytes, including new electrochemical reaction mechanisms, unknown transitional phases formed by diffusion between two solid states and increased contact resistance. The ion diffusion of solid electrolytes is considered as a key factor to determine the kinetics of electrochemical processes.[111] It is reported that ABF and EELS can gain great insight into the solid ion conductor.[112] The increased resistance of all-solid-state batteries was attributed to the surface and interface atomic-level structures revealed by HAADF and ABF in the aberration-corrected STEM.
Aberration-corrected STEM can quickly identify the fundamental chemical and physical causes of problems and figure out feasible ways to solve them. Nowadays, the HAADF and ABF imaging techniques of STEM become powerful in characterizing atomic-level structures of various kinds of materials, which is vital for optimizing the properties of materials and creating new materials. Considering the powerful capability of in situ investigation, in situ STEM has been a focus of the development of STEM. It can provide observation in real time during reaction processes and possibly identify the reaction mechanism. With the development of in situ holders of the STEM, various reactions under different conditions can be implemented in STEM, including heating, cooling and voltage application. However, the interaction between high-energy electron beams and the specimen could also yield undesired electron beam damage to the specimen. To minimize this effect, shorter acquisition time to lower dose and utilization of an accelerating voltage below threshold have been put forward and demonstrated to be effective.[113,114] Finally, a combination of STEM with other probing techniques, including electron energy-loss spectroscopy (EELS) and energy dispersive spectroscopy (EDS), would offer more comprehensive information about various materials. STEM is expected to make more contributions in many fields.





























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}