




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Electronic structures and edge effects of Ga2S2 nanoribbons
[Wang Bao-Ji1, Li Xiao-Hua1, Zhang Li-Wei1, Wang Guo-Dong1, Ke San-Hang2, 3, †,  ]
]
]
|
|


† Corresponding author. E-mail:
Project supported by the National Natural Science Foundation of China (Grant Nos. 11174220 and 11374226), the Key Scientific Research Project of the Henan Institutions of Higher Learning (Grant No. 16A140009), the Program for Innovative Research Team of Henan Polytechnic University (Grant Nos. T2015-3 and T2016-2), and the Doctoral Foundation of Henan Polytechnic University (Grant No. B2015-46).
Ab initio density functional theory calculations are carried out to predict the electronic properties and relative stability of gallium sulfide nanoribbons (Ga2S2-NRs) with either zigzag- or armchair-terminated edges. It is found that the electronic properties of the nanoribbons are very sensitive to the edge structure. The zigzag nanoribbons (Ga2S2-ZNRs) are ferromagnetic (FM) metallic with spin-polarized edge states regardless of the H-passivation, whereas the bare armchair ones (Ga2S2-ANRs) are semiconducting with an indirect band gap. This band gap exhibits an oscillation behavior as the width increases and finally converges to a constant value. Similar behavior is also found in H-saturated Ga2S2-ANRs, although the band gap converges to a larger value. The relative stabilities of the bare ANRs and ZNRs are investigated by calculating their binding energies. It is found that for a similar width the ANRs are more stable than the ZNRs, and both are more stable than some Ga2S2 nanoclusters with stable configurations.
Low-dimensional materials, especially one-dimensional (1D) nanoribbons, have attracted significant attention from the scientific community during the past two decades due to their interesting electronic properties associated with their low dimensionality and the resulting quantum confinement effect. In the past few years, graphene nanoribbons (GNRs), thin strips of graphene, have been extensively studied because of their rich and exotic physical properties which depend on their size and edge termination.[1–3] First-principles calculations have revealed that GNRs with hydrogen saturated armchair- or zigzag-shaped edges always have a nonzero direct band gap which decreases as the ribbon width increases. The band gap variation for armchair GNRs (AGNRs) exhibits distinct “family behaviors”.[4] The band gaps of AGNRs arise from both quantum confinement and the crucial effect of the edges, while for zigzag GNRs (ZGNRs), gaps appear because of a staggered sublattice potential on the hexagonal lattice due to edge magnetization.[1,4–6] Besides GNRs, some other layer-structured nanoribbons, such as boron nitride nanoribbons (BNNRs) and MoS2 nanoribbons, have also been studied intensively.[7–9] It is found that the band gap of hydrogen-terminated zigzag BNNRs is indirect and decreases monotonically with the increasing ribbon width. However, direct band gap oscillation is observed for armchair BNNRs,[10,11] which tends to converge to a constant value when the ribbon is wider than 3 nm. In contrast, zigzag edged MoS2 nanoribbons show a metallic behavior irrespective of the ribbon width, while armchair edged ones are semiconducting and the band gaps converge to a constant value as the ribbon width increases.[12]
As a wide indirect-band-gap semiconductor with uniform layered structure, gallium sulfide (GaS)[13] has been used in photoelectric devices, electrical sensors, and nonlinear optical applications and gained renewed interest.[14] In its bulk form, GaS usually crystallizes into a layered structure, S–Ga–Ga–S (Ga2S2), in which each layer consists of two AA-stacked hexagonal sublayers of Ga atoms sandwiched between two hexagonal sublayers of S atoms. These layers are bound in a three-dimensional (3D) structure by the nonbonding interaction through the S atoms along the vertical axis.[14] Recently, the micromechanical cleavage technique (as originally used in peeling off graphene from graphite) was successfully used to fabricate single-layer sheets of Ga2S2.[15,16] A theoretical calculation based on the density functional theory (DFT) has showed that the single-layer Ga2S2 nanosheet is dynamically stable and is a indirect-band-gap semiconductor with an unusual inverted sombrero dispersion of holes near the top of the valence band.[17] As for the electronic properties of Ga2S2 nanoribbons (Ga2S2-NRs) which are important in realistic device applications, very few reports are available in the literature, to the best of our knowledge.
The purpose of this work is to investigate the electronic structures of Ga2S2-NRs by performing first-principles DFT calculations. The effects of different ribbon widths and different edge structures are studied. Furthermore, the relative stabilities of these nanoribbons are also investigated. In the next section, we describe our computational method. Section 3 presents our calculated results, and the last section is devoted to conclusions.
The models of Ga2S2-NRs are constructed by cutting out a stripe of single-layer Ga2S2 nanosheet with the desired edges and widths. Adopting a similar notation used to describe GNRs,[1,2,18] we use the number of zigzag lines (Nz) or dimer lines (Na) to present the width of a zigzag-edged ribbon (Ga2S2-ZNR) or a armchair-edged ribbon (Ga2S2-ANR) and denote the ribbon by Nz–Ga2S2-ZNR (as illustrated in Fig.
 | Fig. 1. Top and side views of the atomic structures of 6-Ga2S2-ZNR (a) and 11-Ga2S2-ANR (b). The ribbon width and 1D unit cell distance are denoted by Wz (Wa) and dz (da) for the ZNRs (ANRs), respectively. The ribbons are extended periodically along the x direction. |
Calculations of structure optimization and band structure are carried out by using a first-principles pseudopotential plane wave method based on DFT, as implemented in the Vienna ab initio simulation package (VASP).[19] The projector augmented wave method (PAW)[20] is used to describe the ion-electron interaction. The electron exchange and correlation are treated by the generalized gradient approximation (GGA)[21] in the version of Perdew–Burke–Ernzerhof (PBE).[22] A kinetic energy cutoff of 600 eV is adopted for the plane-wave expansion of the wave function. The 4s24p1 electrons of Ga, and 3s23p4 electrons of S are treated as valence electrons. Periodic boundary conditions (PBC) are employed for the infinitely long nanoribbon systems and a vacuum space of 10 Å in each direction perpendicular to the ribbon is used to eliminate the interaction between the periodic images. The atomic structures of the systems, including the lattice parameters and atomic positions, are fully relaxed by the conjugate gradient method until the residual forces acting on each atom are less than 0.001 eV/Å. For the structure optimization a Monkhorst–Packs k-mesh of 1 ×1×12 is used for the Brillouin zone (BZ) sampling. The band structures are presented by 45 k-points along the X axis in the BZ.
We first optimize the atomic structure of a periodic 2D Ga2S2 monolayer, in which Ga–S and Ga–Ga atoms are linked covalently in the quadruple layer. The optimized bond lengths of Ga–S and Ga–Ga are 2.37 Å and 2.47 Å, respectively. The Ga–S–Ga (or S–Ga–S) and Ga–Ga–S bond angles are 100.38° and 117.50°, respectively. The thickness of the quadruple layer is 4.76 Å. The computed binding energy is 3.64 eV per atom. The Ga2S2 monolayer is found to be a semiconductor with an indirect band gap of 2.38 eV on the DFT/PBE level. These results are in good agreement with recent DFT calculations.[17,23] We note that Kohn–Sham DFT calculations usually underestimate band gaps of semiconductors because of the lacking of the derivative discontinuity in the energy functionals. A hybrid DFT calculation[17] gives a larger value of the indirect band gap, 3.28 eV. Although the Kohn–Sham DFT has the band gap issue, it can usually give reasonable shapes of energy bands and wave functions in the vicinity of the Fermi level, compared to results from quasiparticle GW calculations.
We consider two different directions of termination for Ga2S2-NRs, zigzag and armchair, and for each direction of termination we consider the bare and H-saturated edge configurations. In order to investigate the width dependence of the electronic properties, we perform calculations for different ribbon widths, up to Nz = 24 and Na = 19 (i.e., 24-Ga2S2-ZNR and 19-Ga2S2-ANR). Since qualitatively similar results are observed for the different widths, here we first take the 6-Ga2S2-ZNR with a width of 17.37 Å and 11-Ga2S2-ANR with a width of 18.37 Å as prototypes to present our results (see Fig.
Our calculations for the ZNRs show that their electronic structures are spin polarized. To carefully find out the spin configuration of the ground state, we perform total energy calculations for all possible spin configurations: Ferromagnetic (FM), nonmagnetic (NM), and various antiferromagnetic (AFM) configurations. As an example, the results for 6-Ga2S2-ZNR are summarized in Table 1S (see Supplementary Information). These results suggest that the FM configuration is the most stable one for both the bare and H-terminated systems.
The band structures of 6-Ga2S2-ZNR with bare and H-saturated edges are shown in Figs.
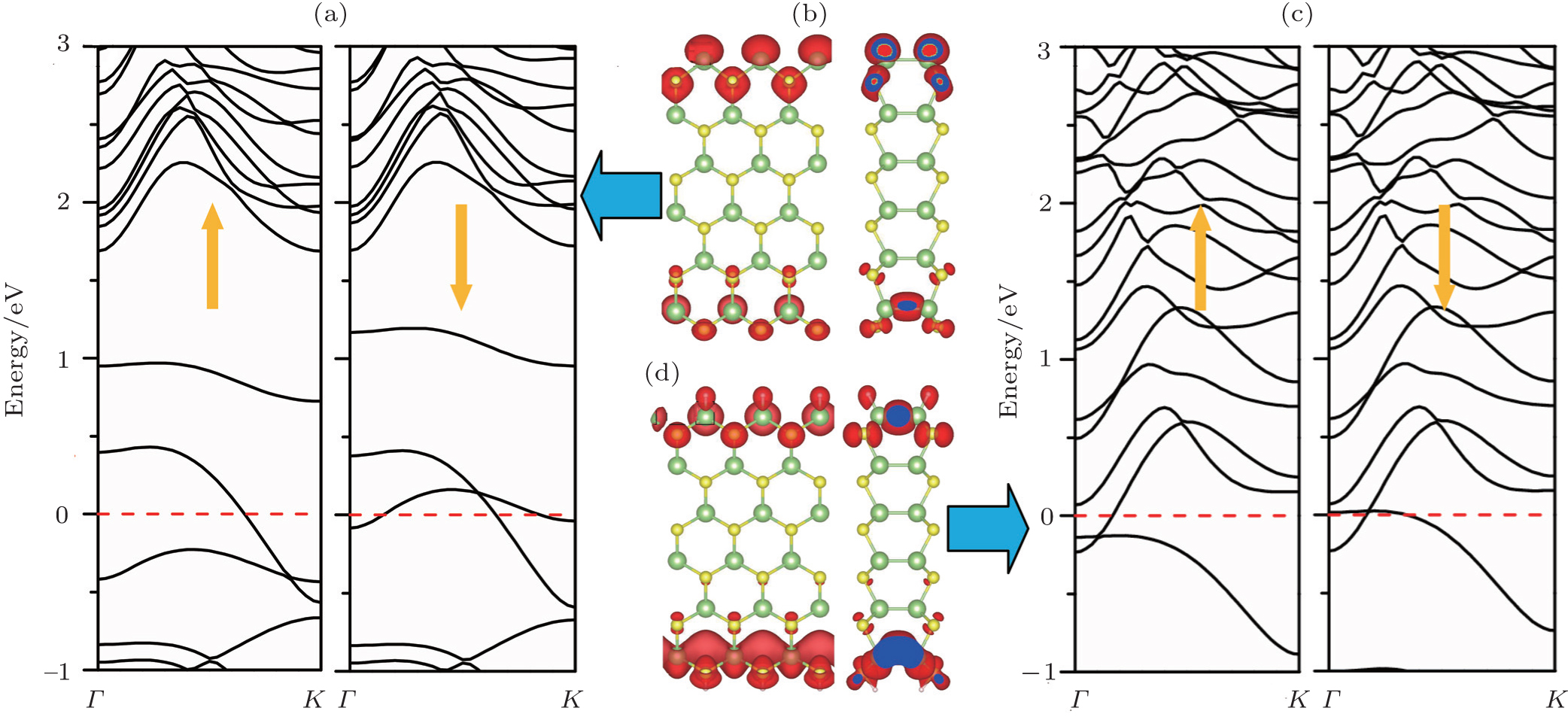 | Fig. 2. Calculated spin-polarized band structures (left: majority spin; right: minority spin) and corresponding local density of states (LDOS) near the Fermi level for bare 6-Ga2S2-ZNR [panels (a) and (b)] and H-terminated 6-Ga2S2-ZNR [panels (c) and (d)]. |
In order to understand the characteristics of these bands near the Fermi energy, we plot their local density of states (LDOS) in Figs.
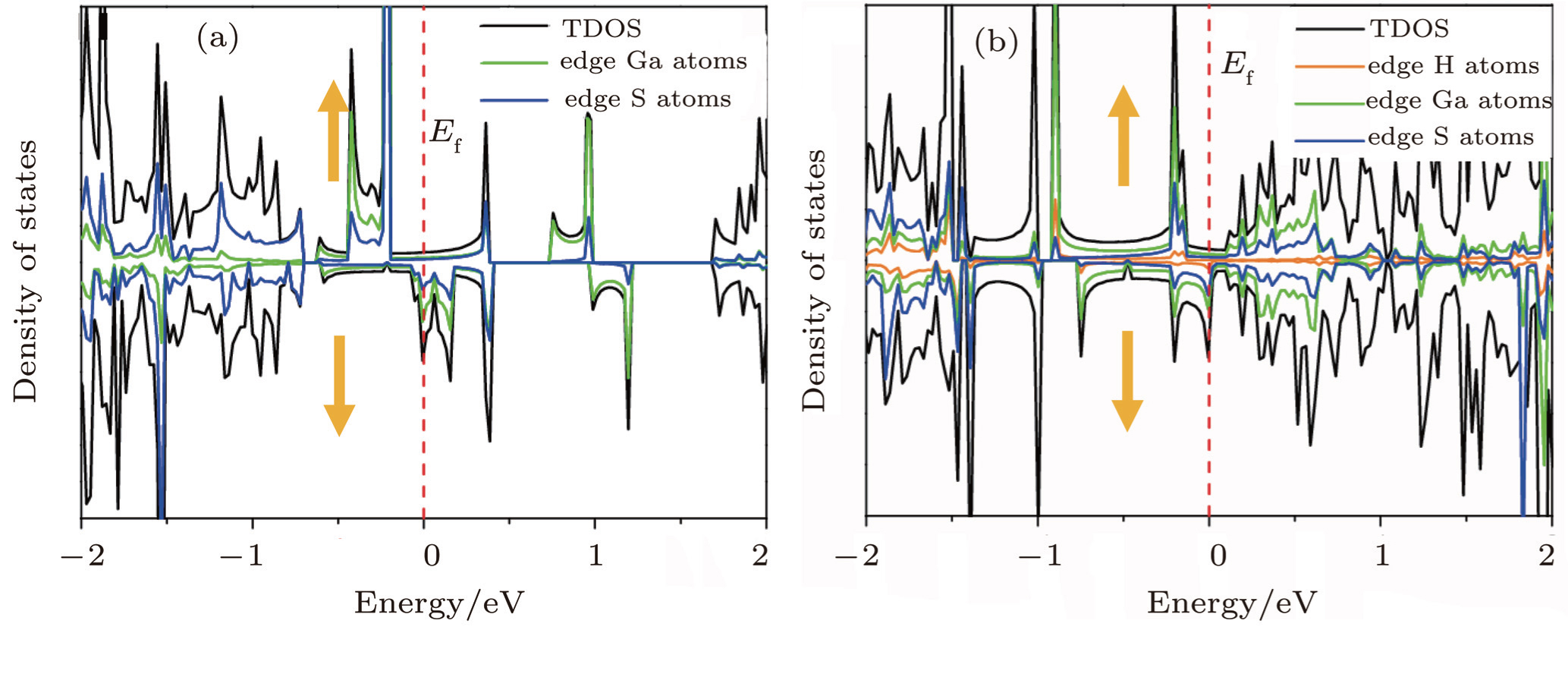 | Fig. 3. Spin-dependent total and partial density of states for bare 6-Ga2S2-ZNR (a) and H-terminated 6-Ga2S2-ZNR (b). |
In contrast, our calculations for the ANRs show that their electronic structures are spin unpolarized. Both the bare and H-saturated Ga2S2-ANRs exhibit a semiconducting nature. As an example shown in Fig.
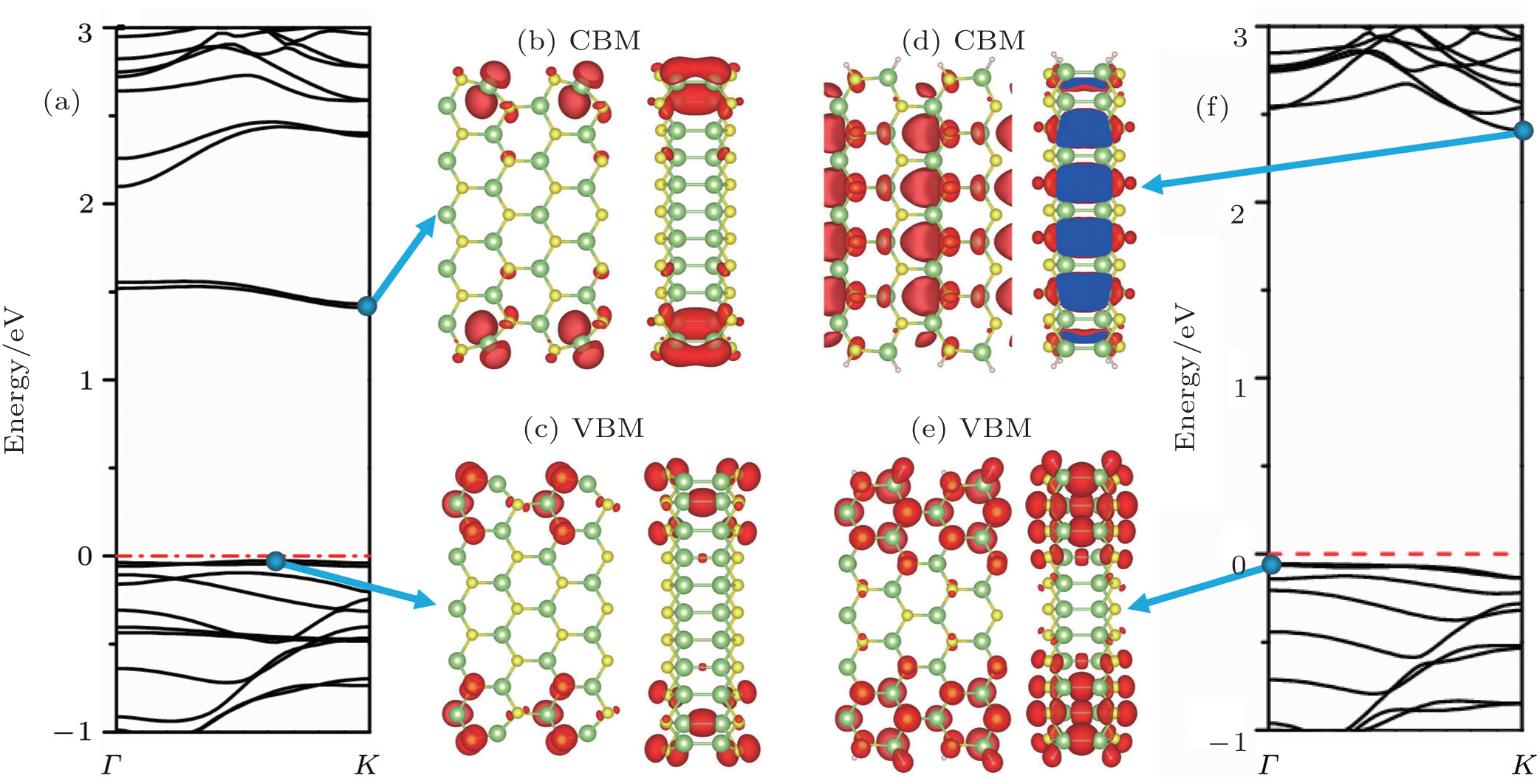 | Fig. 4. Band structures and corresponding LDOS for CBM and VBM of the 11-Ga2S2-ANR. Panels (a), (b), and (c) correspond to the bare Ga2S2-ANR, panels (d), (e), and (f) correspond to the H-terminated Ga2S2-ANRs. Each LDOS figure contains a top view (left) and a side view (right) of the LDOS isosurface of the specified states at CBM or VBM. The Fermi level is set to be zero. |
To analyze the characteristics of the VBM and CBM and the influence of the edges, we calculate the LDOS isosurfaces for the energies around the VBM and CBM of the bare and H-saturated 11-Ga2S2-ANR (see Figs.
When the edge Ga and S atoms are saturated by H atoms, the dangling-bond edge states disappear and thus the band gap increases. As can be seen in Fig.
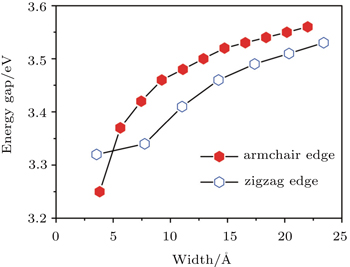
Next let us look at the variation of band gap as a function of ribbon width for both the bare and H-sturated ANRs. As shown in Fig.
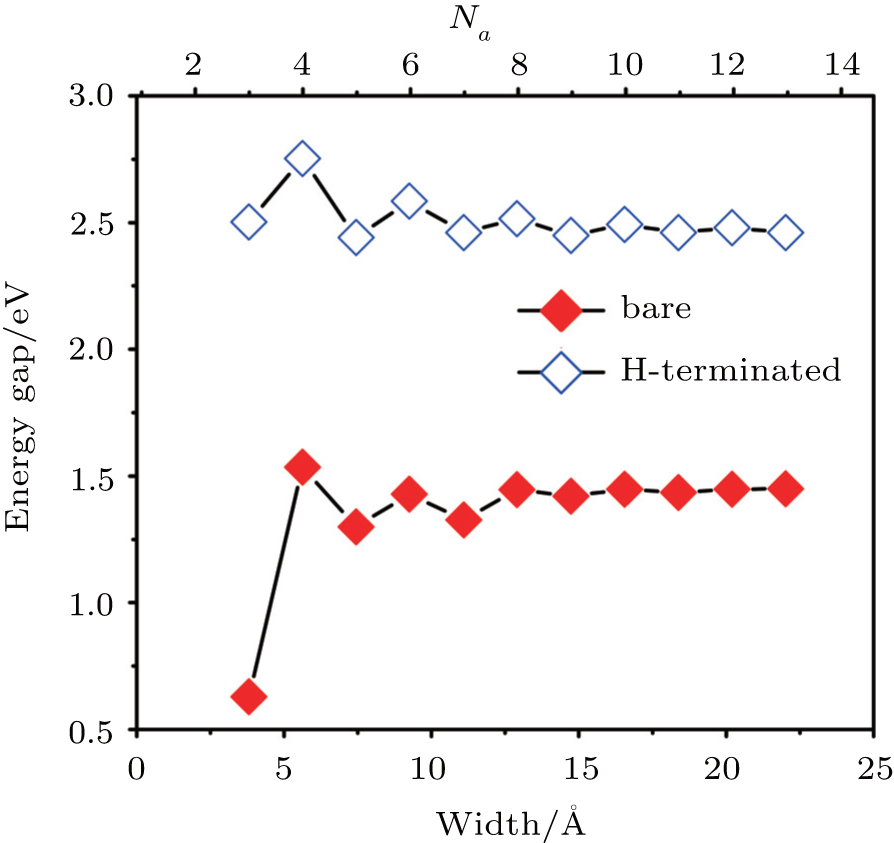 | Fig. 5. Band gaps of a series of bare Ga2S2-ANRs (3 ≤ Na ≤ 13) as a function of the ribbon widths (filled red lozenges). For a comparison, the result for the corresponding hydrogen saturated Ga2S2-ANRs is also shown (voided blue lozenges). |
 | Fig. 6. Binding energy of bare Ga2S2-ZNRs (3 ≤ Nz ≤ 8) and Ga2S2-ANRs (6 ≤ Na ≤ 13) as a function of the ribbon width. |
Finally, we would like to discuss the stability of Ga2S2-NRs, which is quite important because it determines whether this nanostructure can be realized experimentally. The stabilities of different nanoribbons and nanoclusters can be evaluated with their binding energies; those with larger binding energies would be more stable. To estimate the stability, we calculate the binding energy per atom as a function of the ribbon width for bare Ga2S2-NRs in the following way:[12] Eb = (nEGa + mES − EGanSm)/(n + m), where EGa, ES, and EGanSm are the total energies of Ga, S atoms, and GanSm, respectively, with n and m being the number of Ga and S atoms, respectively. As is shown in Fig.
In summary, by performing first-principles DFT calculations we have investigated systematically the electronic properties of Ga2S2 nanoribbons which have not been realized yet but the corresponding 2D monolayer has been successfully fabricated experimentally. We have considered the bare and H-saturated zigzag- and armchair-edged nanoribbons with different ribbon widths (up to 3 nm). It is found that the electronic properties of the nanoribbons strongly depend on their edge structures. The Ga2S2-ZNRs display an intrinsic metallic character with FM coupling, raised from Ga-4p and S-3p states at the Ga-terminated edge while the Ga2S2-ANRs are indirect-band-gap semiconductors without spin polarization. The band gap of bare Ga2S2-ANRs exhibits an oscillation behavior with increasing ribbon width and ultimately converges to a constant value of 1.45 eV when the ribbon becomes wide enough (>13 Å). The H-saturated Ga2S2-ANRs have the similar behavior but with larger converged band gap of 2.46 eV due to the saturation of the dangling bonds at the edges. Finally, we have studied the relative stabilities of the bare ZNRs and ANRs by calculating their binding energy per atom, and found that the ANRs are more stable than the ZNRs with a similar width. The binding energies are found to be slightly smaller than that of the 2D monolayer, but is larger than that of Ga2S2 nanoclusters with stable configurations, indicating that the bare ribbons can be expected to be realized experimentally in the future.
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 | |
| 23 | |
| 24 | |
| 25 | |
| 26 | |
| 27 | |
| 28 | |
| 29 |