

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Graphene-like Be3X2 (X = C, Si, Ge, Sn): A new family of two-dimensional topological insulators
Cite this Article
Song Lingling, Zhang Lizhi, Guan Yurou, Lu Jianchen, Yan Cuixia, Cai Jinming. Graphene-like Be3X2 (X = C, Si, Ge, Sn): A new family of two-dimensional topological insulators. Chinese Physics B, 2019, 28(3): 037101 
Permissions
Graphene-like Be3X2 (X = C, Si, Ge, Sn): A new family of two-dimensional topological insulators
† Corresponding author. E-mail:
Abstract
Using first-principle calculations, we predict a new family of stable two-dimensional (2D) topological insulators (TI), monolayer Be3X2 (X = C, Si, Ge, Sn) with honeycomb Kagome lattice. Based on the configuration of Be3C2, which has been reported to be a 2D Dirac material, we construct the other three 2D materials and confirm their stability according to their chemical bonding properties and phonon-dispersion relationships. Because of their tiny spin–orbit coupling (SOC) gaps, Be3C2 and Be3Si2 are 2D Dirac materials with high Fermi velocity at the same order of magnitude as that of graphene. For Be3Ge2 and Be3Sn2, the SOC gaps are 1.5 meV and 11.7 meV, and their topological nontrivial properties are also confirmed by their semi-infinite Dirac edge states. Our findings not only extend the family of 2D Dirac materials, but also open an avenue to track new 2DTI.
1. Introduction
Two-dimensional (2D) Dirac materials, with a linear band dispersion around the Fermi level and massless carriers that can be described by Dirac equations,[1,2] displaying ballistic charge transport and enormously high carrier mobility, exhibiting special topological phases,[3–11] will offer potential applications for high performance electronic and spintronic devices. However, the Dirac cores in most of these 2D materials can only exist in the absence of spin–orbit coupling (SOC).[12] The SOC will open a gap at the Dirac point and lead to a topological insulating phase, especially when the SOC strength is very strong.[7,13] The 2D topological insulators (TI) exhibit the quantum spin Hall effect with spin-filtered edge states in their bulk SOC gaps.[14,15] In general, if the SOC gap is very small (< 1 meV, e.g., the SOC gap for graphene is about 0.0008 meV),[16] the influence of the SOC will be neglected, and it will be considered that the 2D material is a “real” 2D Dirac material with linear band dispersion around the Fermi level.[17] If the SOC gap is large (> 1 meV), the 2D Dirac materials will be 2DTI.[18,19] Then, the 2D Dirac materials are more likely composed of light elements (e.g., B, C, N) and the 2DTI are more likely constructed by heavy elements (e.g., Sn, Pb, Bi). Up to now, lots of 2D Dirac materials (or 2DTI) have been predicated and investigated, which include graphene,[1] silicene,[18,20] germanene,[18,20] several kinds of graphynes,[21–23] topological insulators on semiconductor surfaces,[4–6,9–11] 2D organic TI,[6–8] and so on. Especially for graphene, as the only 2D Dirac material that has been truly confirmed in experiment, it has attracted intense interest over the past decade.[1,24]
Graphene, graphynes, and silicene are all single-element materials. Compare to these single-element materials, bi-element materials have more complex structures and more potential to be modulated. Up to now, lots of bi-element 2D Dirac materials and 2DTI have been predicted and investigated, e.g., polyaniline (C3N),[25] C4N-I,[25] C4N-II,[25] TiB2,[26] and MC6 (M = Mo, W).[27] Recently, Wang et al. have predicted a new 2D Dirac material, monolayer Be3C2, by using the global particle-swarm optimization method and density functional theory.[28] We know that the group IV elemental monolayers have played a crucial role in the field of 2D materials, where graphene (C) is a Dirac material, silicene (monolayer Si),[18,20] germanene (monolayer Ge),[18,20] and stanene (monolayer Sn) are topological insulators.[29] Then, what is the stability and electronic properties of the Be3X2 (X = Si, Ge, Sn)?
In this work, by using first-principle calculations, we investigated the stability and electronic properties of honeycomb Kagome (HK) mixed 2D materials Be3X2 (X = C, Si, Ge, Sn), and found that all of them are 2DTI. More interestingly, monolayer Be3C2 and Be3Si2 have very tiny SOC gaps (< 1 meV), which means that both are 2D Dirac materials. According to the semi-infinite Dirac edge states of these four materials, we can confirm that Be3Ge2 and Be3Sn2 are 2DTI, and their SOC gaps are much larger than those of Be3C2 and Be3Si2.
2. Computational details
The density-functional calculations are performed by using the plane wave basis Vienna ab initio simulation pack (VASP).[30] The projector-augmented plane wave (PAW) approach is used to represent the ion–electron interaction.[31] The Perdew–Burke–Ernzerhof (PBE) generalized gradient approximation is adopted for the electron–electron interactions.[32] The energy cutoff of the plane wave is set to 600 eV with a precision energy of 10−4 eV/atom and a precision force of.01 eV/Å. The Monkhorst–Pack scheme with 15×15×1 and 95×95×1 k-points is used to sample the Brillouin zone for structural optimization and self-constant electronic properties calculation, respectively. In all the calculations, the 18 Å vacuum layer is used to avoid the spurious interactions between periodic images. To assess the dynamical stability, phonon dispersions are calculated based on the finite displacement method which is embedded in the phonopy program.[33,34] The semi-infinite Dirac edge states are calculated by using the Wannier90 package.[35,36]
3. Results and discussion
3.1. Geometrical properties and structural stabilities
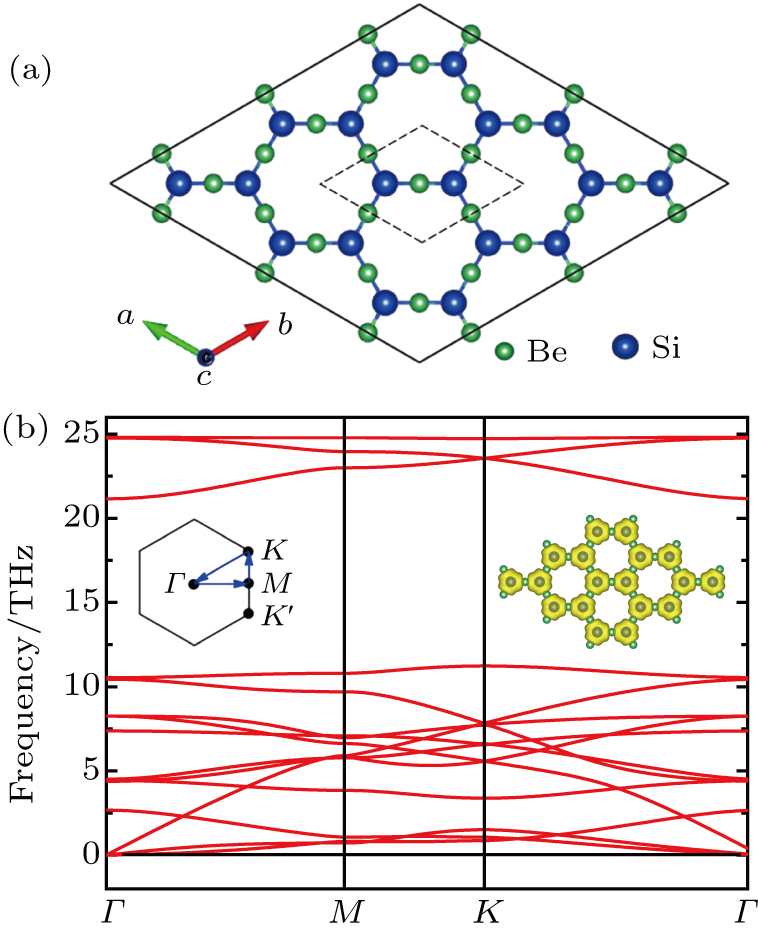
A geometrical configuration of monolayer Be3Si2 is shown in Fig.
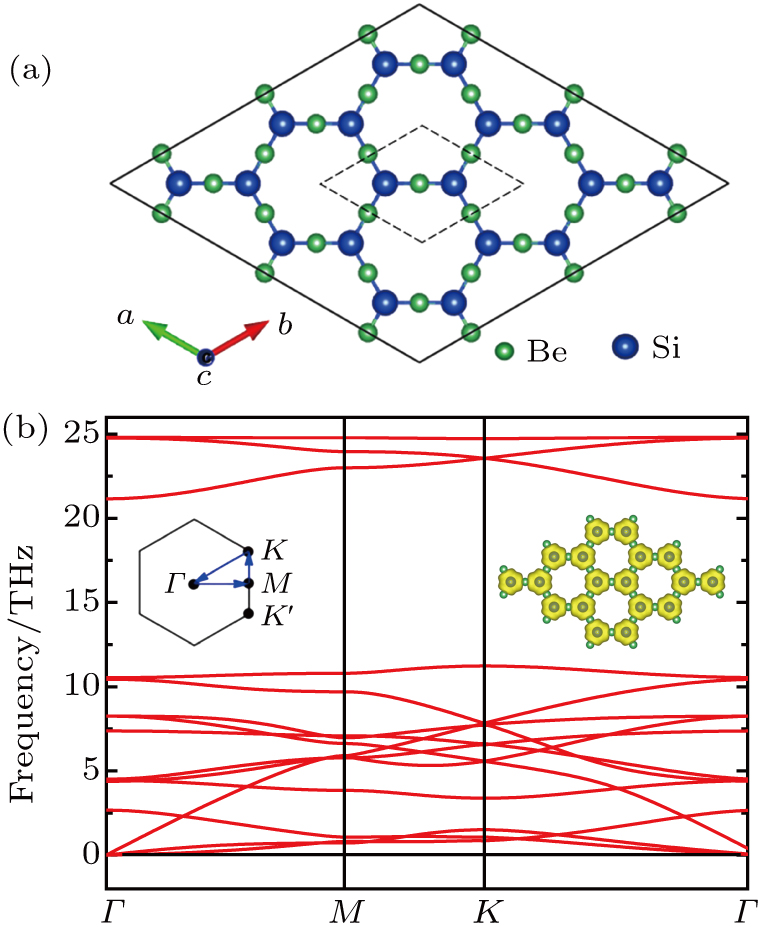 | Fig. 1. (a) Optimized configuration of Be3Si2 monolayer with a unit cell labeled by black dashed line. The green and blue spheres represent Be and Si, respectively. (b) Phonon-dispersion curves, where the left inset indicates the first Brillouin zone, and the right inset shows the electron localization function of the Be3Si2 monolayer with the iso-value of 0.96. |
| Table 1. Lattice constants a, band gaps within SOC Eg, and Fermi velocities VF of graphene, silicene, and Be3X2 . |
3.2. Electronic properties and Dirac states of Be3Si2
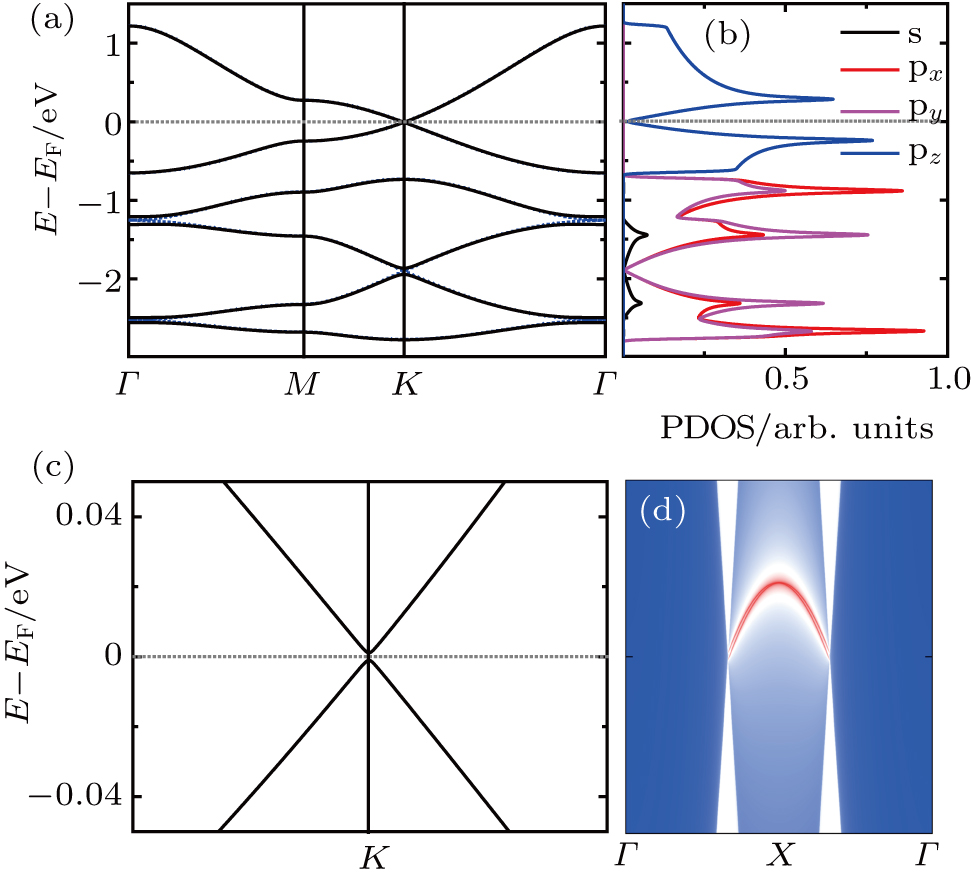
To investigate the electronic properties of monolayer Be3Si2, we calculate its band structures and corresponding projected density of states (PDOS), as shown in Fig.
 | Fig. 2. Electronic structures of Be3Si2: (a) band structure and PDOS, (b) 3D valence and conduction bands around the Fermi level, (c) and (d) the real space charge distributions of HOB and LUB. |
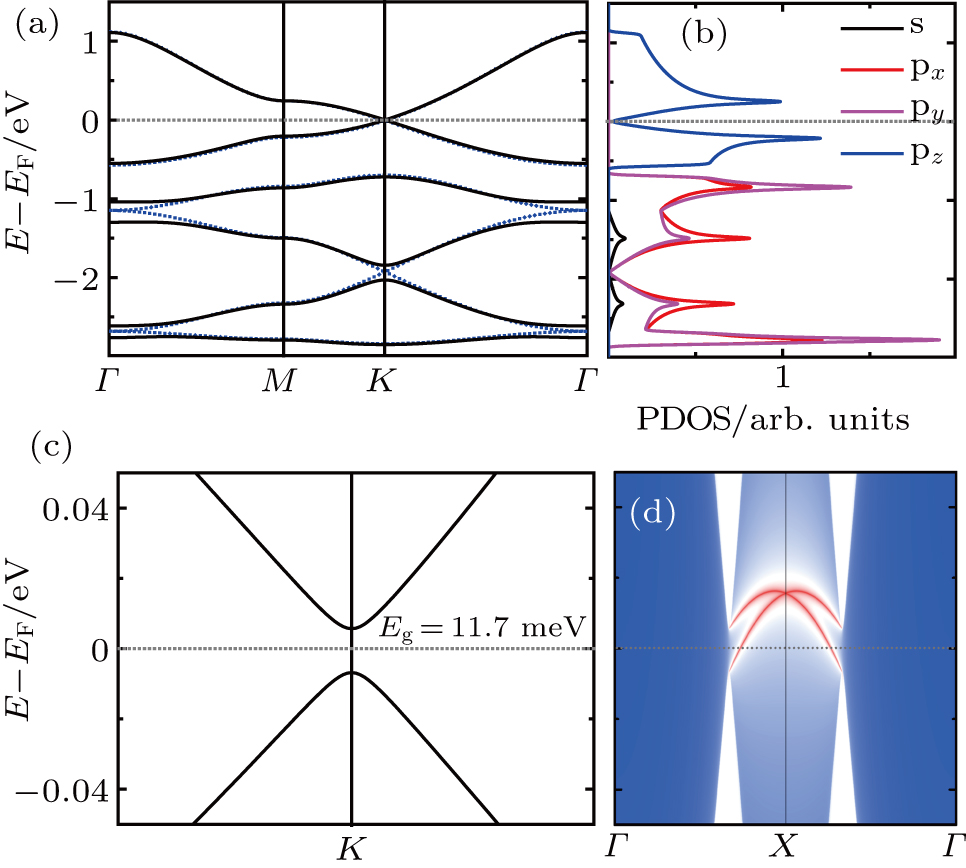
Mostly, Dirac cores can only exist in the absence of SOC.[12] But Be3C2 can be considered as 2D Dirac materials even with SOC, because its SOC gap is very small (0.04 meV).[28] For Be3Si2, the SOC can open a tiny band gap of 0.06 meV, being also much smaller than 1 meV, which means that Be3Si2 is one new “real” 2D Dirac material. We know that silicene is a 2DTI with a SOC band gap about 1.55 meV.[18] The large SOC gap comes from buckled properties of silicene, where some of the in-plane orbitals contribute to the bands around the Fermi level.[20] Here, there is no buckle for Be3Si2, and only the pz orbitals (out of plane orbitals) of Si contribute to the bands around the Fermi level. Similar to graphene and Be3C2, Be3Si2 has a very small SOC gap, which leads it to be a 2D Dirac material.
3.3. Topological properties of Be3X2
In Fig.
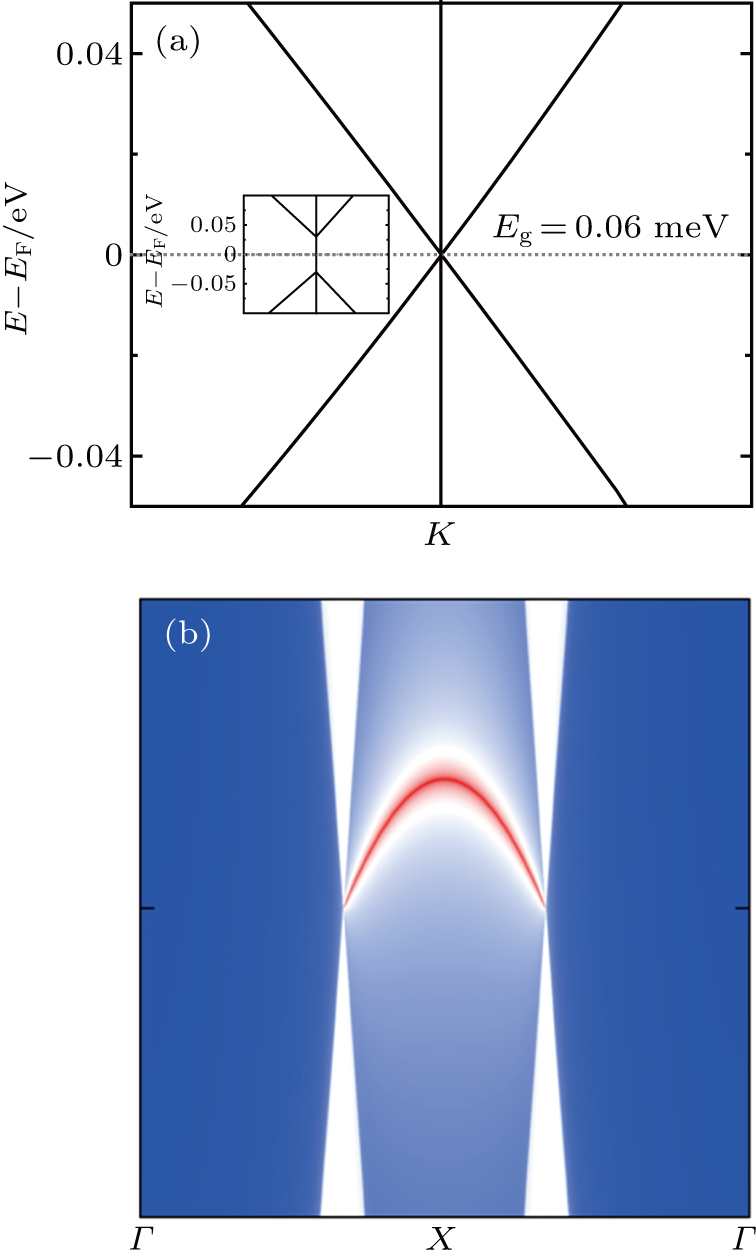 | Fig. 3. (a) Band structures of Be3Si2 with SOC around K points, where the inset is the zoom-in view around the Fermi level. (b) The semi-infinite Dirac edge states around the Fermi level. |
 | Fig. 4. Electronic properties of Be3Ge2. (a) Band structures with (black line) and without (blue dash) SOC. (b) Projected density of states without considering SOC. (c) Zoom-in figure around K points of the band structures considering the SOC. (d) The semi-infinite Dirac edge states around the Fermi level. |
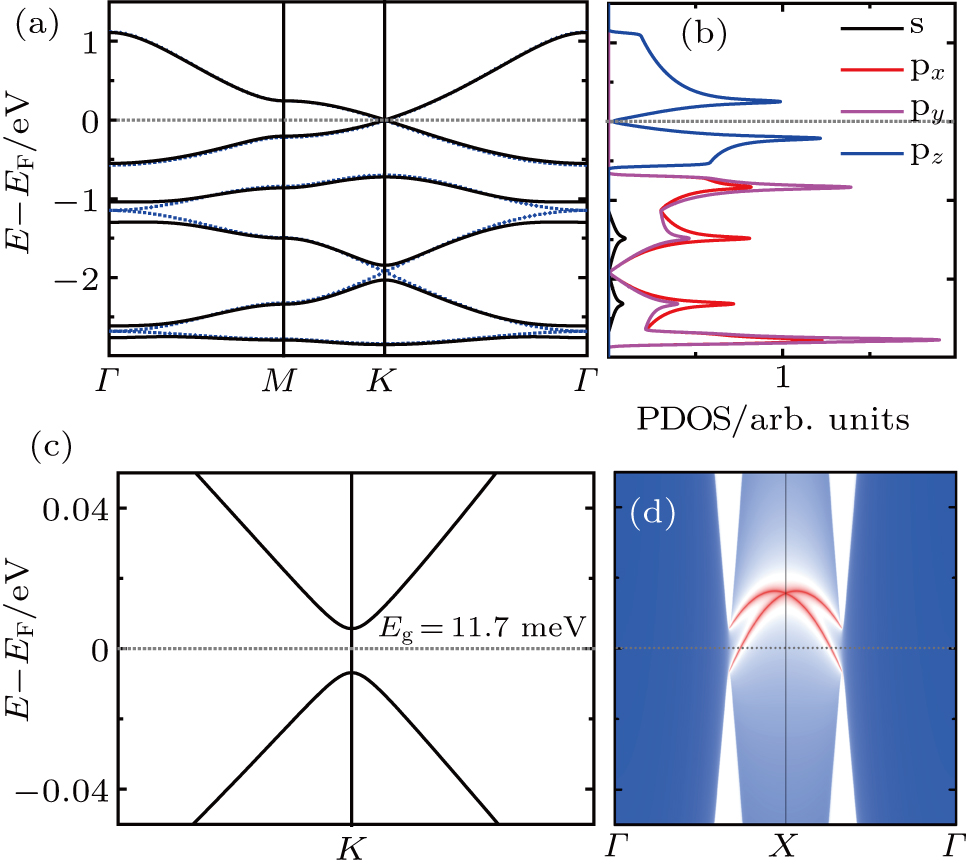 | Fig. 5. Electronic properties of Be3Sn2. (a) Band structures with (black line) and without (blue dash) SOC. (b) Projected density of states without considering SOC. (c) Zoom-in figure around K points of the band structures considering the SOC. (d) The semi-infinite Dirac edge states around the Fermi level. |
More interestingly, for Be3Sn2, its deep energy bands exhibit much larger SOC band gaps (∼0.2 eV) and semi-infinite Dirac edge states (Fig. S2). These large SOC band gaps mean that we can realize a large gap 2DTI if we move the Fermi level to these gaps by hole doping.
4. Conclusion
We investigate a family of 2DTI with HK lattice, monolayer Be3X2 (X = C, Si, Ge, Sn). Based on the chemical bonding properties and phonon-dispersion relationships, we have confirmed their stabilities. By investigating their electronic and topological properties, we find that Be3C2 and Be3Si2 can be considered as 2D Dirac materials because of their tiny SOC band gaps. For Be3Ge2 and Be3Sn2, the SOC gaps are 1.5 meV and 11.7 meV, and their topological nontrivial properties are also confirmed by their semi-infinite Dirac edge states. Since the atomic numbers of the Ge and Sn atoms are larger than those of the C and Si atoms, the spin–orbit coupling effect is larger. Our research will enrich the family of 2DTI and promote the development of 2D materials.
Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] | |
| [33] | |
| [34] | |
| [35] | |
| [36] | |
| [37] | |
| [38] | |
| [39] |