{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A computational study of the chemokine receptor CXCR1 bound with interleukin-8
Cite this Article
Wang Yang, Severin Lupala Cecylia, Wang Ting, Li Xuanxuan, Yun Ji-Hye, Park Jae-hyun, Jin Zeyu, Lee Weontae, Tan Leihan, Liu Haiguang. A computational study of the chemokine receptor CXCR1 bound with interleukin-8. Chinese Physics B, 2018, 27(3): 038702 
Permissions
A computational study of the chemokine receptor CXCR1 bound with interleukin-8
† Corresponding author. E-mail:
Abstract
Abstract
CXCR1 is a G-protein coupled receptor, transducing signals from chemokines, in particular the interleukin-8 (IL8) molecules. This study combines homology modeling and molecular dynamics simulation methods to study the structure of CXCR1-IL8 complex. By using CXCR4-vMIP-II crystallography structure as the homologous template, CXCR1-IL8 complex structure was constructed, and then refined using all-atom molecular dynamics simulations. Through extensive simulations, CXCR1-IL8 binding poses were investigated in detail. Furthermore, the role of the N-terminal of CXCR1 receptor was studied by comparing four complex models differing in the N-terminal sequences. The results indicate that the receptor N-terminal affects the binding of IL8 significantly. With a shorter N-terminal domain, the binding of IL8 to CXCR1 becomes unstable. The homology modeling and simulations also reveal the key receptor-ligand residues involved in the electrostatic interactions known to be vital for complex formation.
1. Introduction
Chemokines are a group of proteins that regulate a large number of biological and pathological processes, such as promoting embryogenesis, angiogenesis, wound healing, and as a guide in signaling, playing important roles in both innate and adaptive immune systems.[1,2] Chemokine achieves a plethora of its activities by activating G-protein coupled receptors (GPCR) on the cell surface.[3–6] Upon binding to the GPCRs, chemokine initiates a conformational change, which activates signaling pathways. In humans, there are two types of high affinity chemokine receptors, CXCR1 and CXCR2, that have been identified to interact with the chemokine interleukin-8 (IL8). The binding of the IL8 to CXCR1 plays vital roles in various signal transduction pathways. In tumor progression and metastasis, CXCR1 has been identified as a target for blocking the formation of breast cancer stem cells and malignant melanoma that drives both tumor growth and metastasis.[7] The binding of IL8 to CXCR1 induces a conformational change to the receptor, activating the G-protein subunit, and the subsequent release of the subunits. Therefore, it is importance to understand the exact interactions between IL8 and CXCR1 in structural aspects.
The structure of IL8 is the first chemokine structure that has been solved.[8–10] IL8 exists both as a monomer and dimers in solution, and a monomeric IL8 alone binds to CXCR1 with high affinity.[11,12] The structure of IL8 is composed of a helix, three anti-parallel strands, and a C-terminal helix. The structure of IL8 contains two conserved disulfide bonds. One is linked between C7 and C34 and the other is linked between C9 and C50. The N-terminal located near C7 and C9 is approximately 10 amino acids in length, which has been considered as a crucial binding region.[13–17] The N-terminal of IL8 was thought to initially bind to the transmembrane (TM) domain of receptor CXCR1 mediated by electrostatic interactions before settling to its final position on the extracellular pocket. A series of NMR experiments, mutagenesis, and simulation studies have provided crucial insights into the binding of IL8 to CXCR1.[18–25] These research works focused on the interaction between the N terminal of CXCR1 and IL8 and found that the residues located at N terminal of CXCR1 influences the binding of IL8 to CXCR1.
The experimental studies on binding of IL8 to CXCR1 have thus far focused on the extracellular domain of the CXCR1. Charged residues located near extracellular loop region (ECL) and TM domain have been reported to be important for IL8 binding and IL8-mediated signal transduction, while the cysteine residues at the extracellular side are critical for the overall folding of the receptor.[26,27] In 2012, the NMR structure of CXCR1 in DMPC bilayers (PDB ID: 2LNL) was solved,[28] which paved the way for further molecular dynamics study on the CXCR1.[29] In both CXCR1 and the homologous receptor CXCR4, charged residues are mainly located near the membrane and extracellular interface. The negatively charged residues are clustered in the extracellular loops. In addition, four charged residues D85 (TM2), K117 (TM3), D288, and E291 (TM7) form a cluster in the core of the helical bundle of CXCR1. One of these residues (D288) is not conserved in CXCR4 and may contribute to the differences in biological activities of two chemokine receptors.
Although the three-dimensional structure of CXCR1 has been determined by using NMR spectroscopy, the complex structure of the CXCR1 with IL8 has not been experimentally resolved. Based on the structure of the homologous receptor, CXCR4, that is in a complex with a viral chemokine vMIP-II (PDB code: 4RWS),[30] we used a computational modeling approach to build homology models of CXCR1-IL8 complex. The CXCR4-vMIP-II complex has two binding sites, one is in the extracellular domain near the N terminal of the receptor and the other is the orthosteric binding site near the extracellular interface of the receptor, common in most class A GPCRs. The viral chemokine was shown to interact in the first binding site. The available NMR structures of CXCR1 and the CXCR4-vMIP-II complex provide the opportunity to study the CXCR1-IL8 complex computationally. This study focuses on the binding modes and receptor–ligand interactions between CXCR1 and IL8. The results reveal the detailed interactions at the binding sites for the CXCR1-IL8 complex and explain the crucial role of CXCR1 N-terminal residues in the complex formation.
2. Methodology
2.1. Homology modeling
By using homology modeling, four models of the CXCR1-IL8 complex were constructed (see Fig.
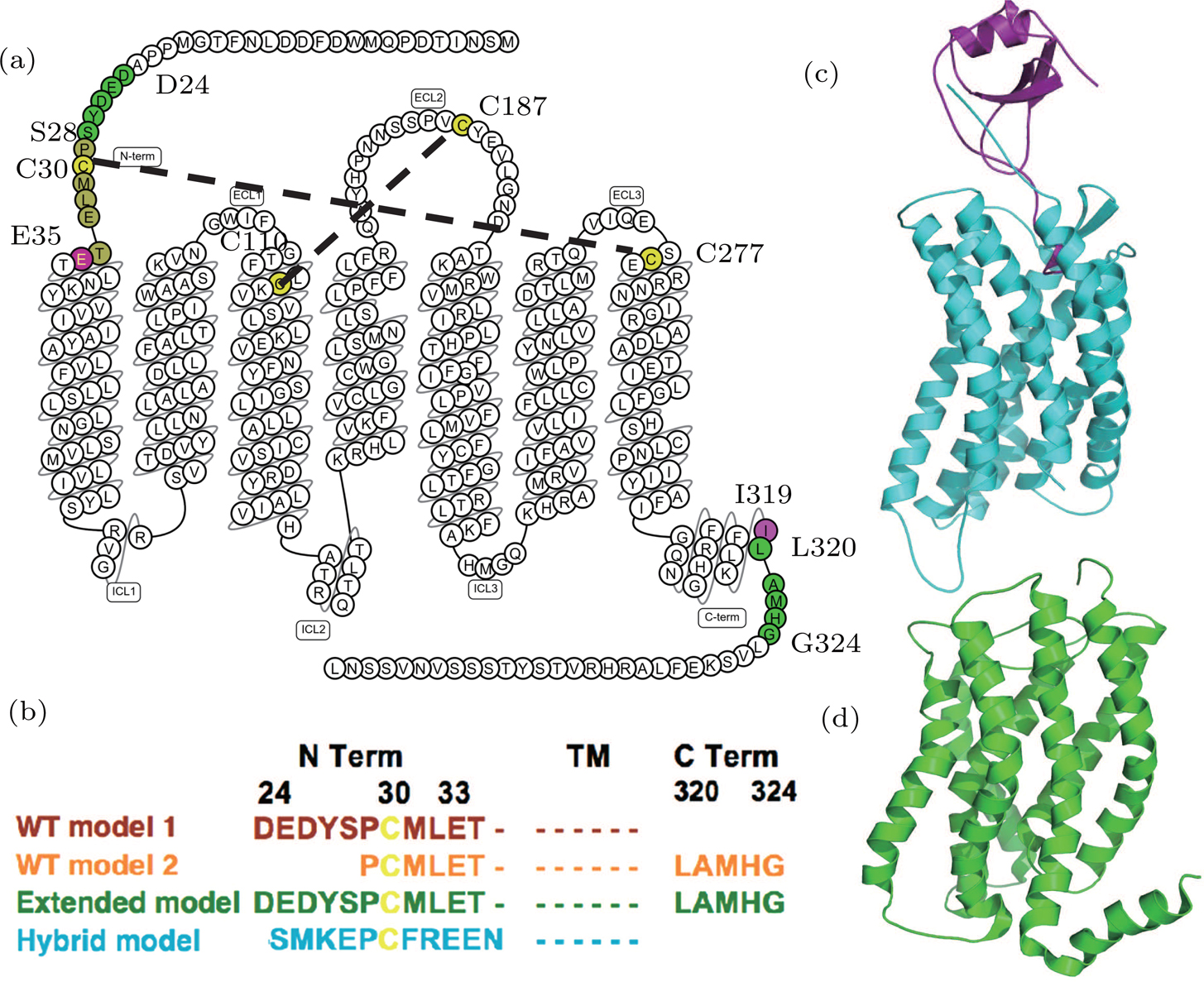
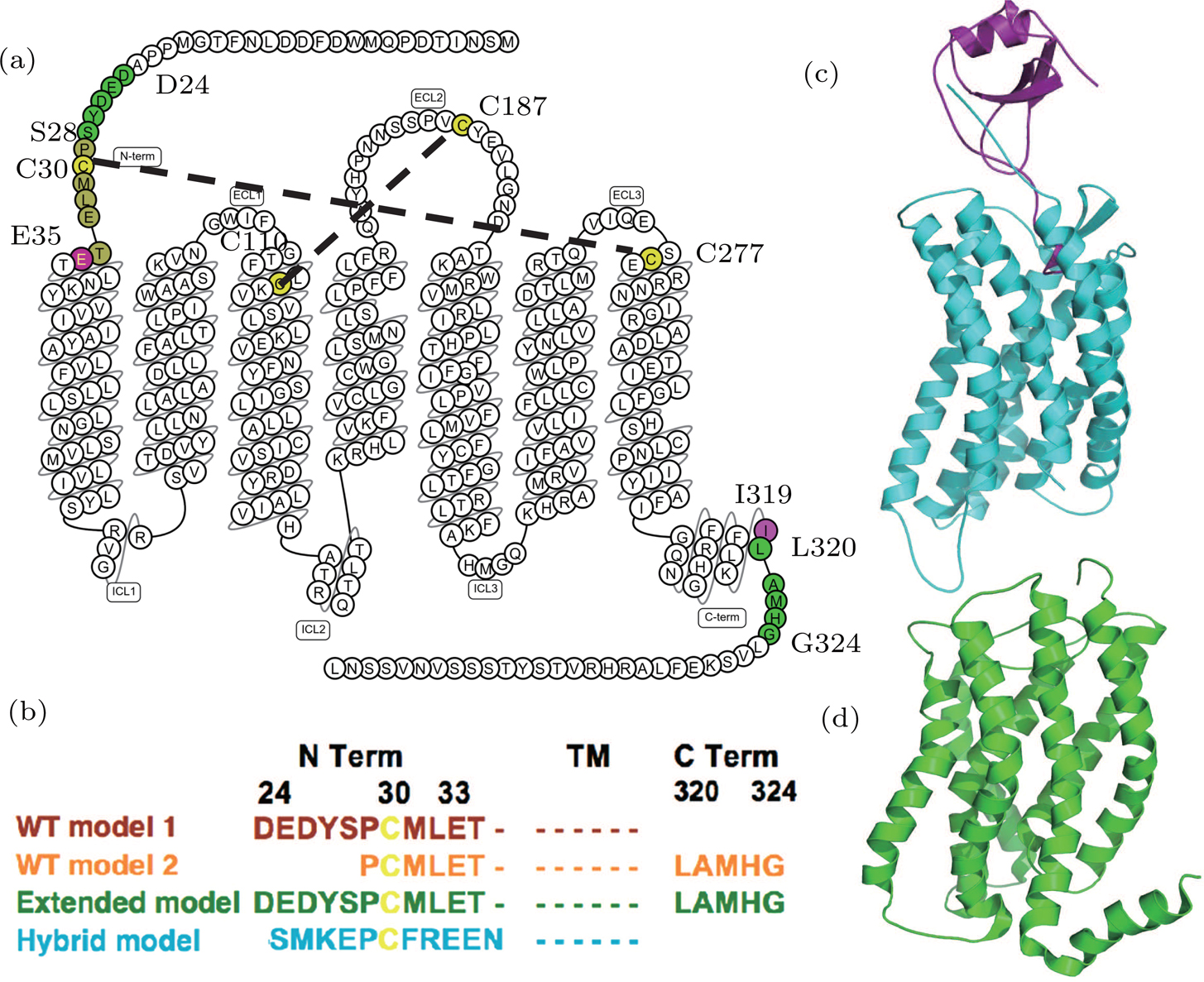 | Fig. 1. (color online) The CXCR1-IL8 systems. (a) The sequence and secondary structure of CXCR1. The snake map is drawn from GPCR db.[32] The residues (C30, C110, C187, and C277) that form disulfide bonds are colored in yellow. The sequence between two purple residues E35 and I319 forms the main body of the CXCR1 receptor. (b) Sequence differences of four constructed models. The four models are colored in brown (WT model 1), orange (WT model 2), green (extended model), and cyan (hybrid model), respectively. (c) The crystal structure of CXCR4-vMIP-II binding complex (PDB code: 4RWS). (d) The NMR structure of CXCR1 (PDB code: 2LNL). |
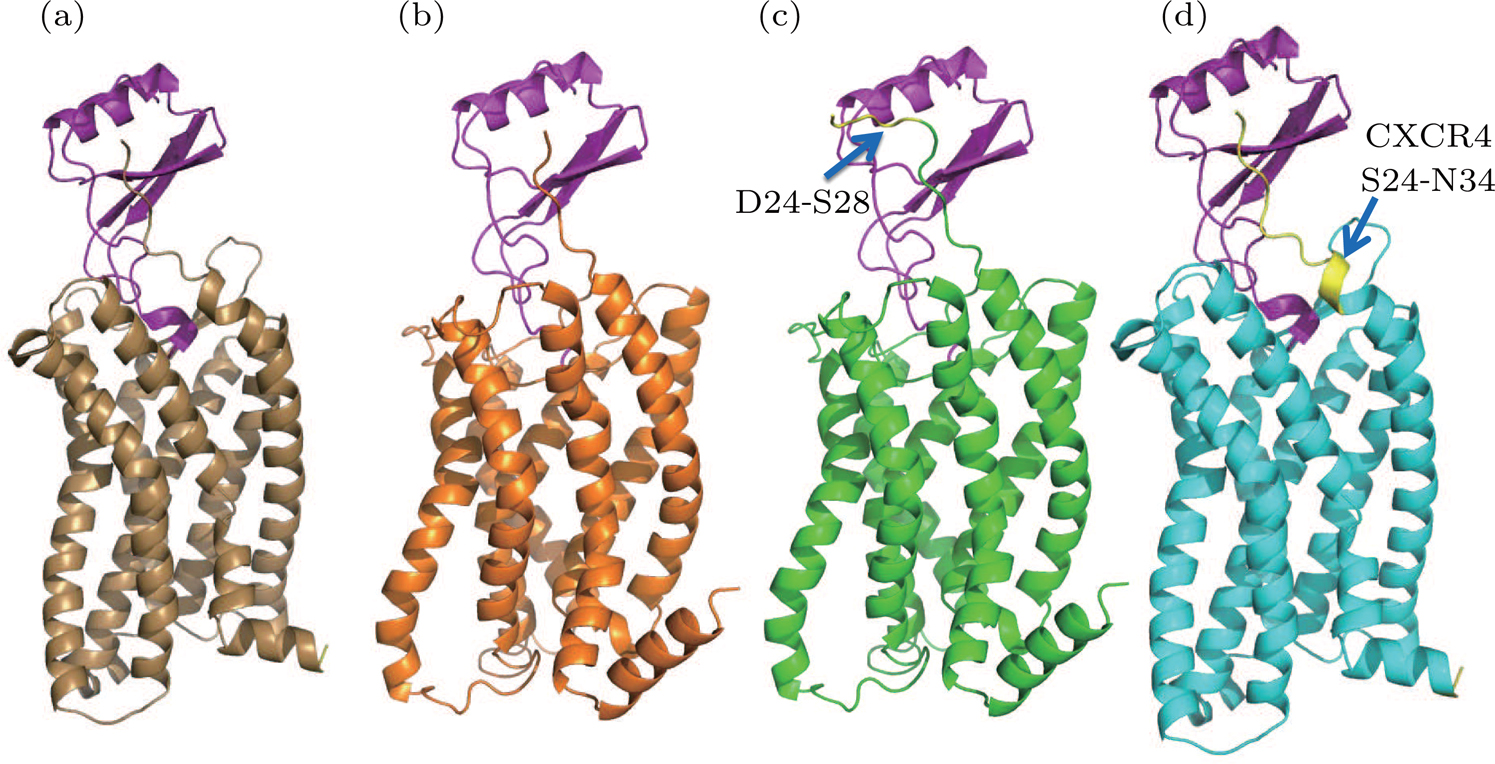
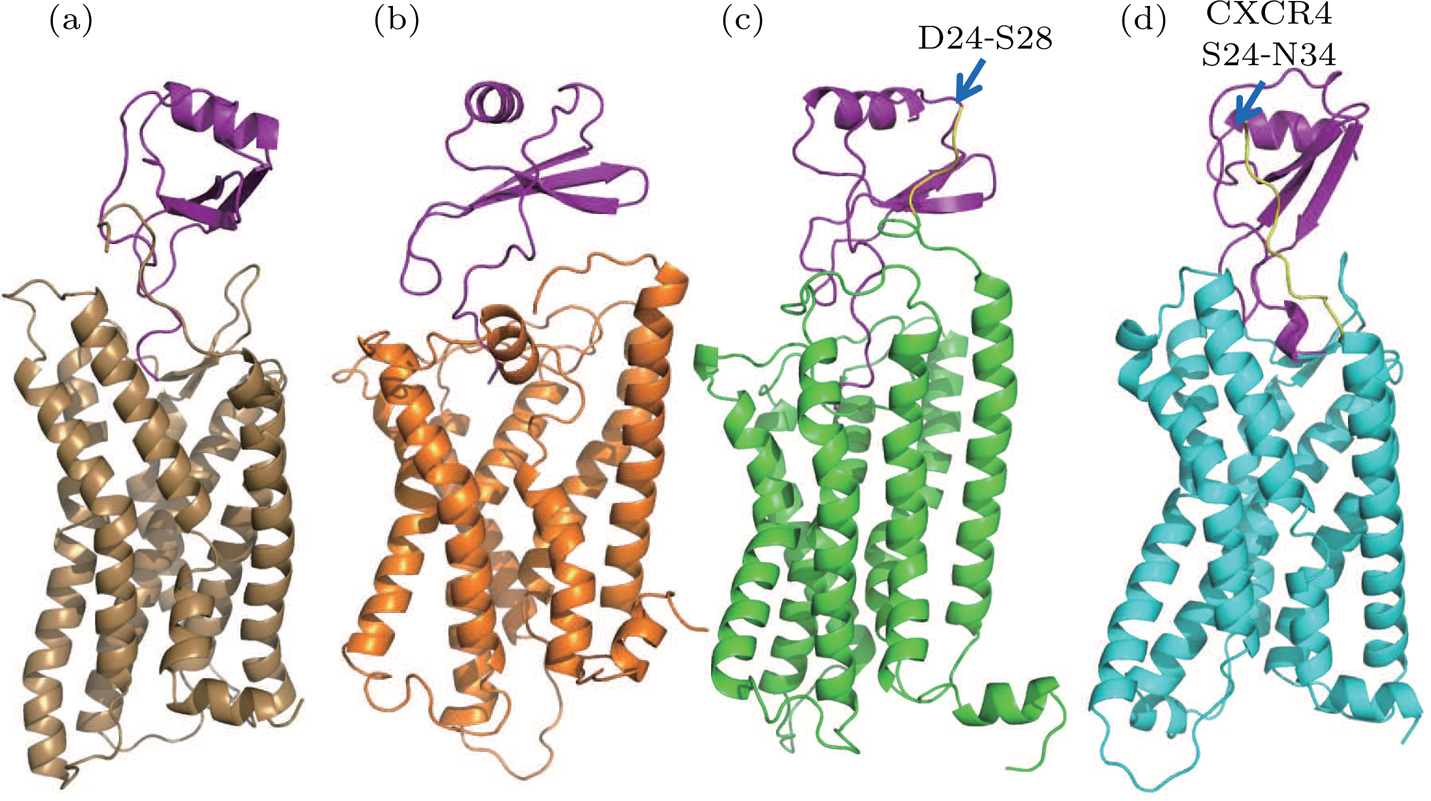
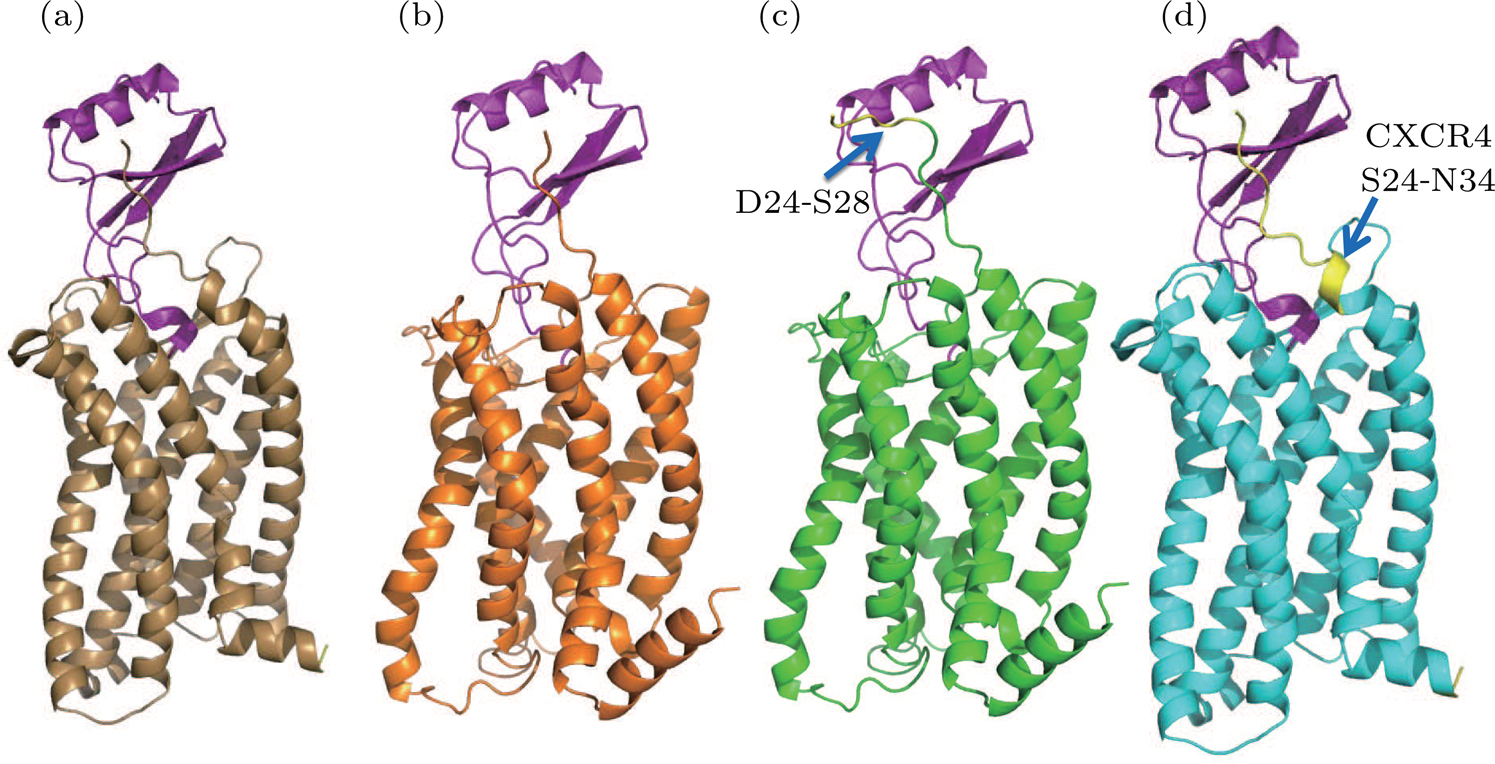 | Fig. 2. (color online) The predicted structures using homology modeling method. (a) WT model 1 (brown). (b) WT model 2 (orange). (c) Extended model (green). (d) Hybrid model (cyan). The residues in N terminal for the extended model and hybrid model are highlighted by yellow color and indicated by the blue arrows. |
The crystallography structure of CXCR4-vMIP-II indicated two binding sites between chemokine and its receptors. The binding site-I was the N terminal of CXCR1 interacting with the ligand IL8. The orthosteric binding pocket of the receptor was referred to as binding site-II. IL8 and vMIP-II have about 21% sequence similarity; while the CXCR1 and CXCR4 have about 38% similarity for full sequence and 42% for TM regions. CXCR4-vMIP-II complex was used as the template for the homology modeling of CXCR1-IL8 complex. Homology models were built by using the I-TASSER server using specified templates.[31]
Wild type (WT) homology model 1 The CXCR4-vMIP-II structure was used as the template. The receptor was modelled based on the CXCR4 structure and the IL8 was modeled according the vMIP-II structure, the binding pose was similar to the CXCR4-vMIP-II. From the target–template sequence alignment, amino acids D24-I319 of CXRC1 were included to build the receptor in this model.
WT-model 2 Different from WT-model 1, the NMR structure of CXCR1 was aligned to the CXCR4-vMIP-II complex to form a virtual CXCR1-vMIP-II complex structure, which was then used as the template to build this model.
The major difference between WT-model 2 and WT-model 1 is at the termini due to the sequence alignment between template and target. The N terminal of model 2 (using the NMR-CXCR1 structure as the template) was five residues shorter (D24-S28) than the counterpart of model 1 (CXCR4-vMIP-II structure as the template), while the C terminal of model 2 was five residues (L320-G324) longer than the counterpart of model 1. This sequence difference is shown in Fig.
Extended model To investigate the role of N terminal in the binding of IL8, another homology model was constructed based on the WT model 2 by extending the N-terminal to include the five missing residues (D24-S28) compared to the WT model 1 (see Fig.
Hybrid model Based on WT-model 1 (the CXCR4-vMIP-II complex template), the hybrid model was constructed by grafting the N-terminal of CXCR4 (S24 to N34) to the WT-model 1. During model construction, the I-TASSER server was instructed to build CXCR1 structure for the sequence of E35-I319 and the N terminal residues of CXCR4 were added manually to the resulting model (i.e., WT-model 1).
2.2. Molecular dynamics simulations
The PPM server was used to reorient the CXCR1-IL8 systems to ensure that the trans-membrane parts of CXCR1 or CXCR4 were well located in the DMPC (1,2-dimyristoyl-sn-glycero-3-phosphocholine) lipid bilayer.[33] For each system, 500 ns all-atom molecular dynamics (MD) simulations were performed. Before the production simulations, the systems were equilibrated in a DMPC lipid bilayer and solvated in a water box. CHARMM-GUI was used to generate topology and parameter files for CXCR1-IL8 systems.[34] In addition to the protein complex, 250 DMPC molecules, about 17000 water molecules, and excess sodium chloride ions were added to maintain an ion concentration of 150 mM; thus, the entire system contained about 90000 atoms. The systems were modelled using the force field parameters of CHARMM36.[35] The NPT ensemble (constant pressure and constant temperature) MD simulations were generated using GROMACS 5.1.2.[36] The REDUCE program in AMBER was used to add hydrogens to the original PDB files and determine the protonate state of histidine residues.[37,38]
The initial energy minimizations were achieved using the steepest descent algorithm, followed by a two-stage equilibration, a 20 ns NVT (constant particle number, volume, and temperature) dynamics with large restraint force constants (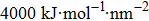
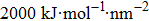
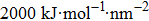
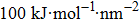
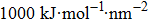
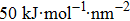
2.3. Analysis
To evaluate the stability of CXCR1-IL8 complex, the structure changes of the homology models were quantified using the root mean square deviation of atomic position (RMSD) of the TM bundle of the receptor with respect to the initial models. For the binding position of ligand IL8, the RMSD of IL8 was computed using the transformation matrix obtained by aligning the TM bundle of the receptor. This described the changes of relative motion of the ligand. The CPPTRAJ package in AMBER tools was used to compute the 2D RMSD of the ligand-receptor complex to check the overall stability of the complex structure.[39] For the 2D RMSD case, the RMSD was calculated between pairwise snapshots obtained from the 500 ns simulation trajectories. We used RMSD 2 Å as the difference cutoff to define structure clusters. In other words, if the RMSD value is smaller than 2 Å, the two structures were clustered in one group. The clustering results were used to gauge the stability of the ligand-receptor complex. The root mean square fluctuations (RMSF) at residue levels in the ligand and receptor were calculated to compare the thermal fluctuations of the systems. Finally, the final structures of all the homology models were studied to compare the binding modes, and the key interactions such as salt bridges between CXCR1 and IL8 were extracted from the simulation results. We used the distance 4 Å as the distance cutoff to search possible salt bridge pairs between the ligand and receptor that formed within 500 ns simulation for each model.
3. Results
3.1. Stability and binding mode of the homology models
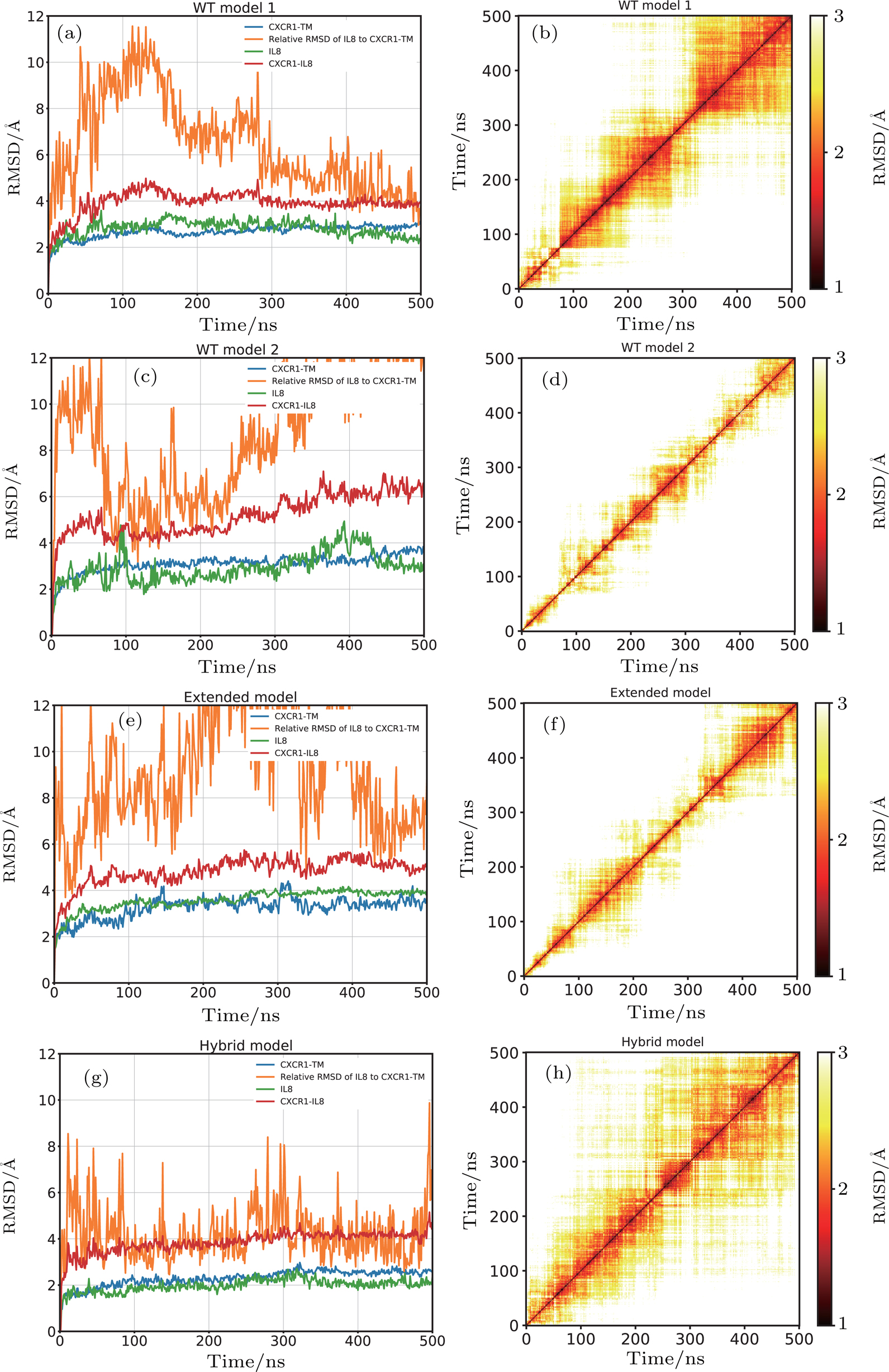
For WT-model 1, the RMSD of the CXCR1 and IL8 with respect to their initial structures were below 3 Å, which suggested that the structures of CXCR1 and IL8 were stable (Fig.
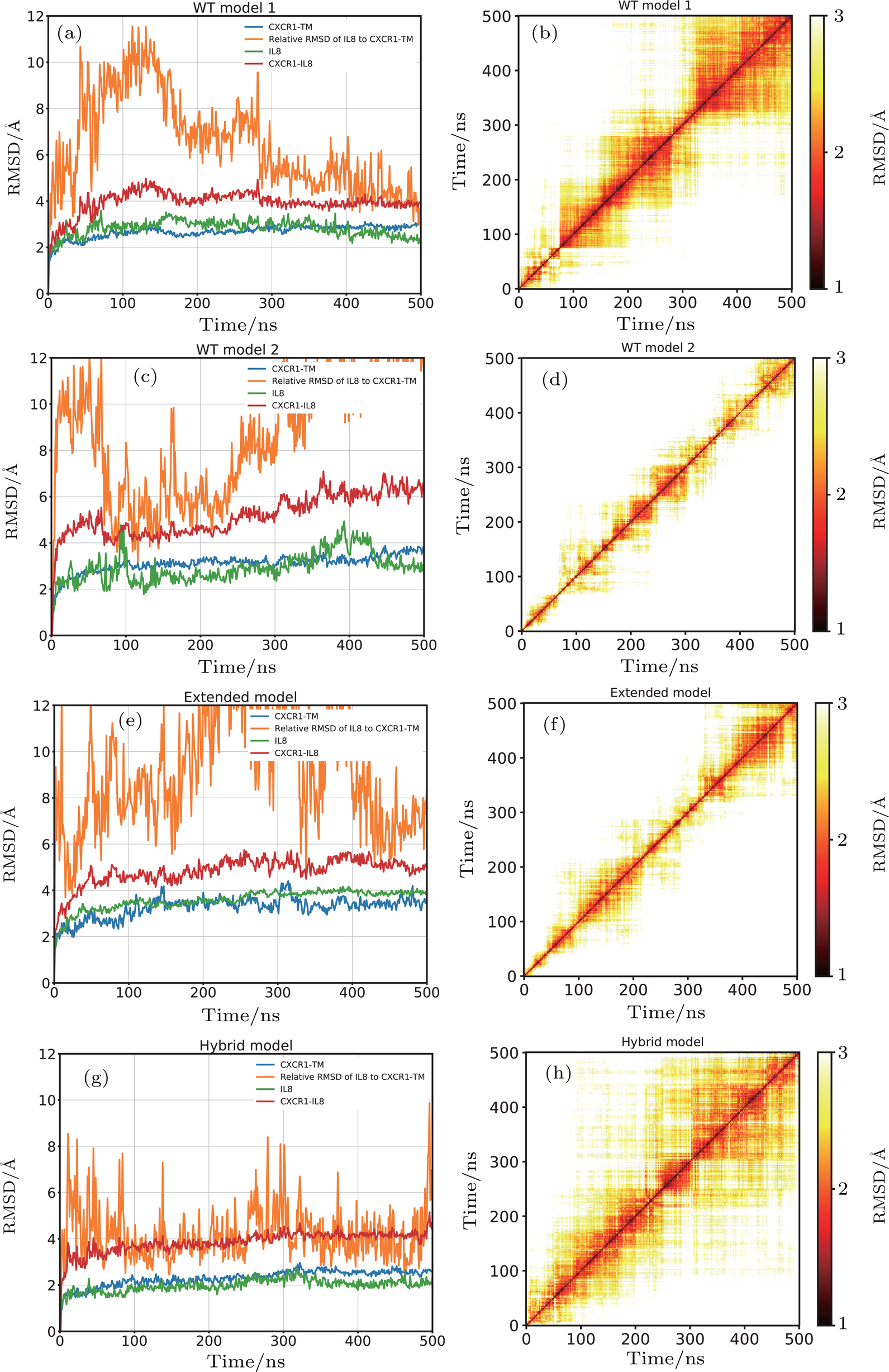 | Fig. 3. (color online) The stability of the CXCR1-IL8 complexes in four different models. (a), (c), (e), (g) The RMSD with respect to the initial structure for CXCR1 TM domain (blue), RMSD of IL8 after aligning the complex with respect to the initial structure of CXCR1 TM bundle domain (orange), RMSD of IL8 with respect to the initial structure of IL8 (green), and RMSD of the overall CXCR1-IL8 complex (red). (b), (d), (f), (h) The 2D-RMSD plot for pairwise conformation comparison of the overall complex. (a), (b) WT model 1; (c), (d) WT model 2; (e), (f) extended model; and (g), (h) hybrid model. |
For WT-model 2, the CXCR1 and IL8 subunits exhibited a similar behavior, revealed by the subunit RMSD with respect to their initial structure, fluctuating around 3 Å (Fig.
In the case of the extended model, the RMSD values of the CXCR1 and IL8 subunits were stabilized around 3 Å, similar to the wild type models. The relative RMSD of IL8 after fitting the CXCR1 TM bundle converged to ∼6 Å, suggesting that the positional difference IL8 relative to CXCR1 compared to the initial model is small (Fig.
For the hybrid model, the RMSD values of the CXCR1 TM and IL8 subunits were the lowest compared to the other three models, indicating more stable subunits in this case. The relative movement of IL8 with respect to CXCR1 was also smaller throughout the 500 ns simulation. The relative RMSD of IL8 after aligning to CXCR1 was below 6 Å most of the time, suggesting tighter binding of IL8 to the CXCR1 (Fig.
3.2. Thermal fluctuations of the predicted complex structures
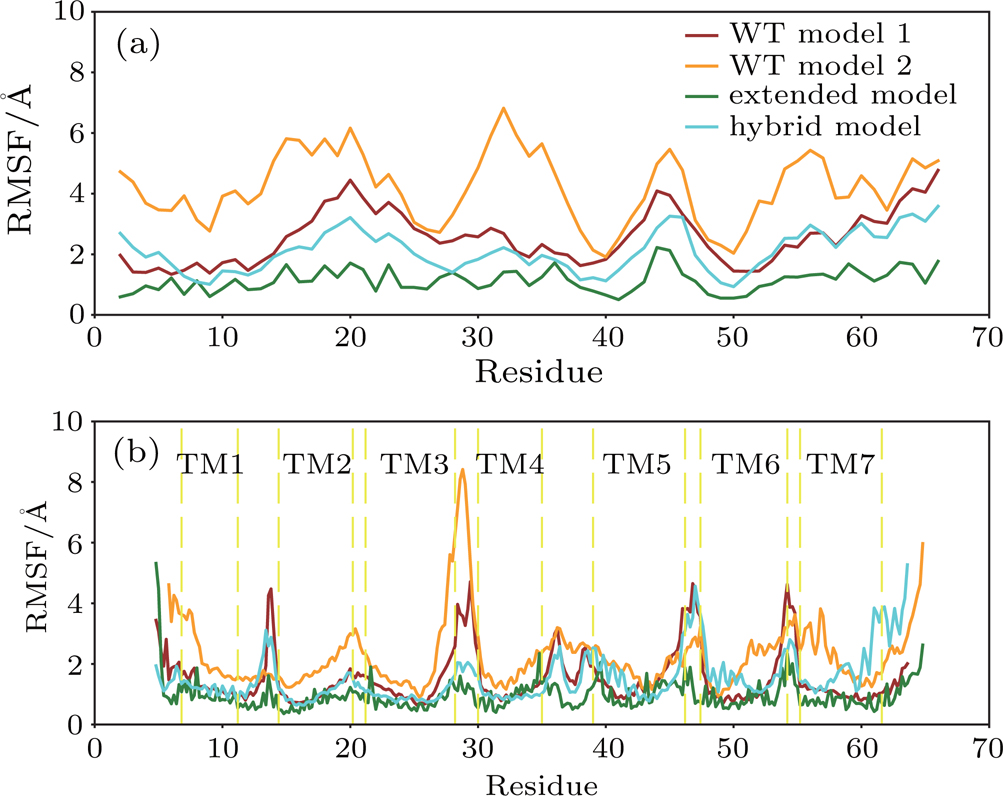
The thermal fluctuations of the complex were quantified using the RMSF at residue levels. The comparison of RMSF for both IL8 and CXCR1 in four models is shown in Fig.
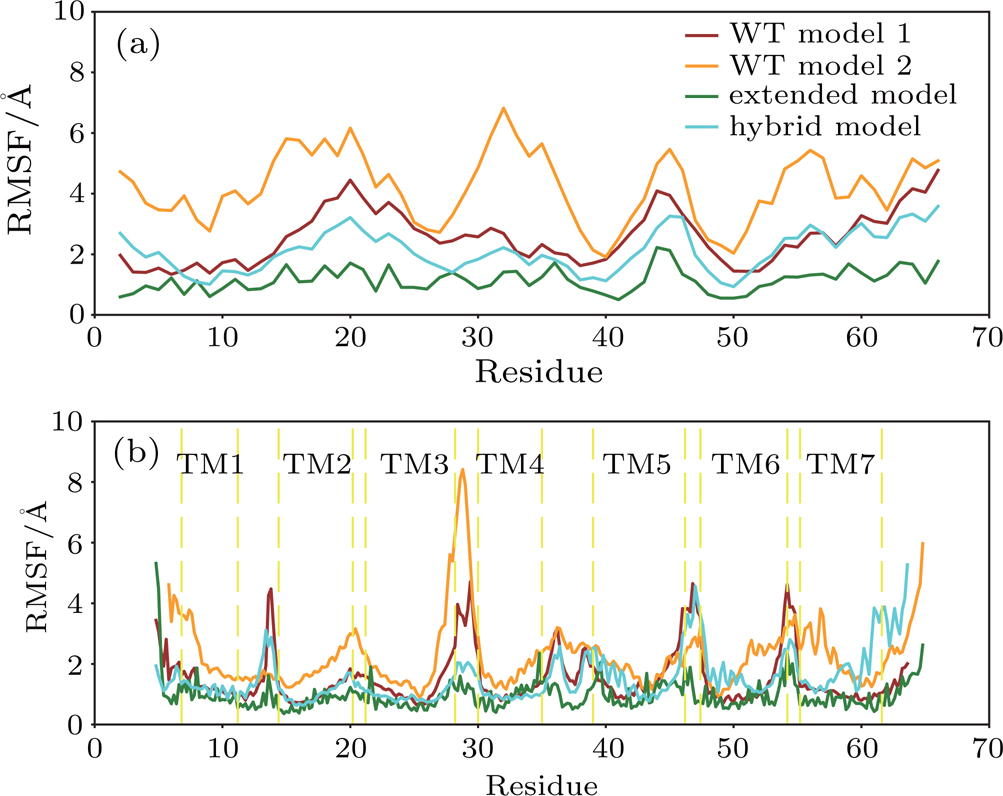 | Fig. 4. (color online) Thermal fluctuation of the predicted models at residue levels. The RMSF of IL8 (a) and CXCR1 (b) for WT model 1 (brown), WT model 2 (orange), extended model (green), and hybrid model (cyan). |
The RMSF value of IL8 and CXCR1 for the extended model was much smaller than that of WT model 2, despite large scale structural rearrangement (see Fig.
3.3. The key residues for the CXCR1-IL8 complex formation
The final structures of the WT model 1, WT model 2, extended model, and hybrid model are depicted in Fig.
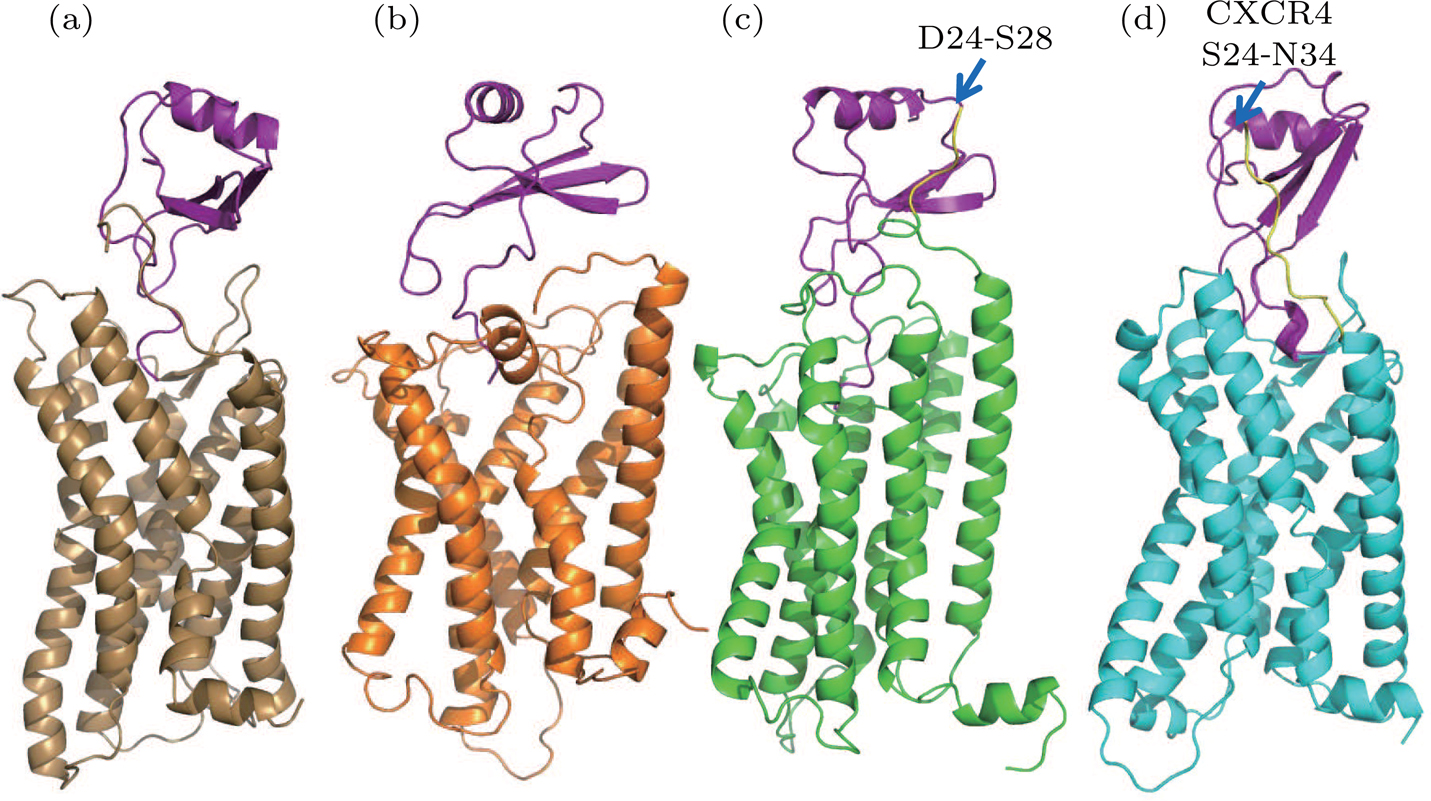 | Fig. 5. (color online) Final structures obtained from simulations: (a) WT model 1 (brown), (b) WT model 2 (orange), (c) extended model (green), (d) hybrid model (cyan). The extended residues and hybrid residues in N terminal are colored in yellow. |
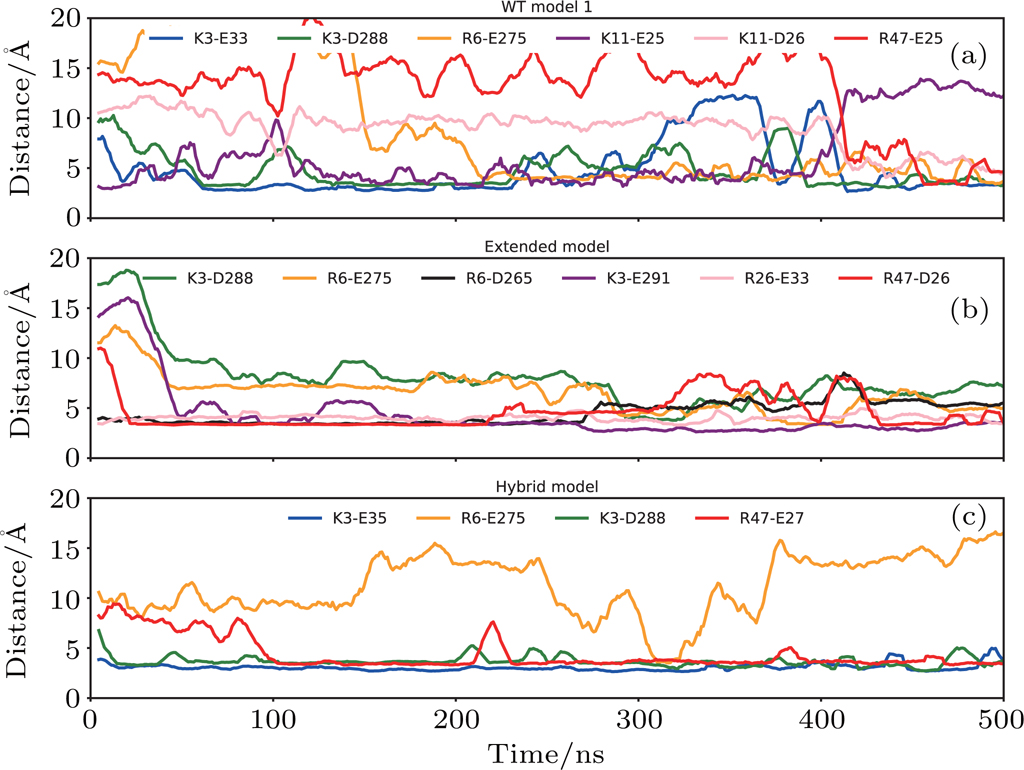
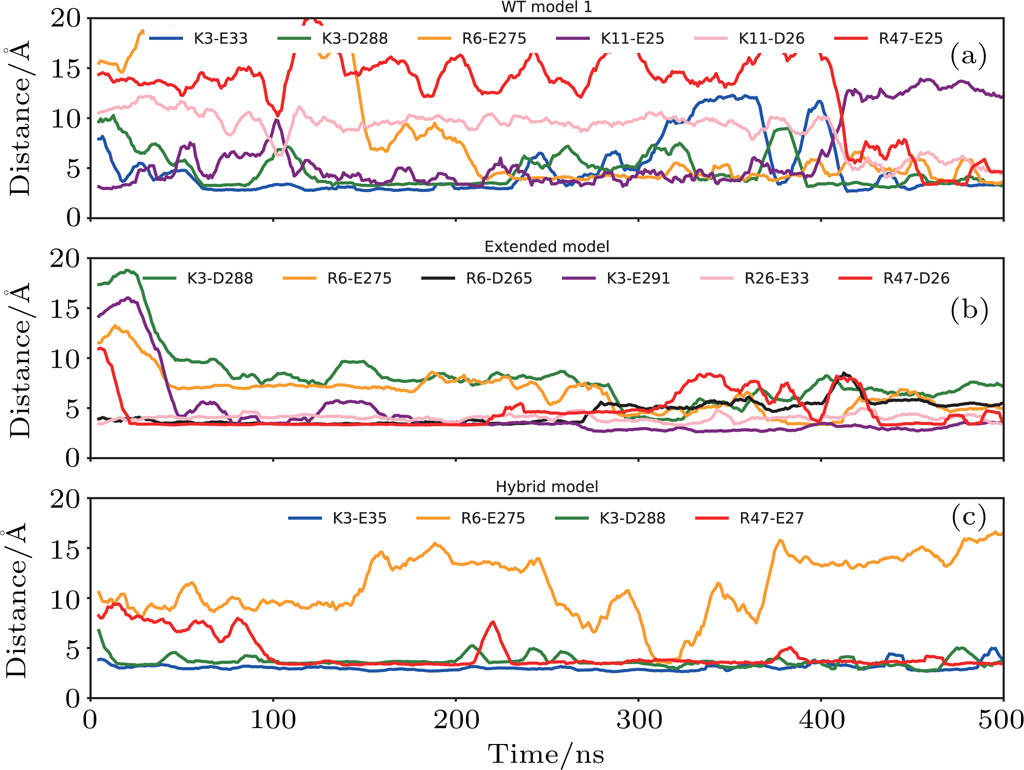 | Fig. 6. (color online) The progression of distances between residues of salt bridge partners: (a) WT model 1, (b) extended model, (c) hybrid model. The distance–time curve was smooth by the window of 10 ns. |
| Table 1.
Stable salt bridge between IL8 and CXCR1. . |
In the case of WT model 2, because the N terminal of CXCR1 was modelled shorter than WT model 1, the favorable interaction between IL8 and CXCR1 at the binding site I could not be established, and the rotation of IL8 around the Z-axis during the simulation indicated that the N-terminal of CXCR1 was required to lock the IL8 in a specific binding position. The large relative RMSD of IL8 (orange curve in Fig.
The binding was improved in the case of the extended model where the N terminal of CXCR1 was extended so that the interactions with IL8 at binding site I could be maintained. The salt bridges between D26/E33 of CXCR1 and R26/R47 of IL8 were observed in binding site I, meanwhile the N terminal of IL8 located in the binding pocket of CXCR1 also formed stable interactions at the binding site II (Fig.
For the hybrid model, the binding of IL8 to CXCR1 was stabilized by the salt bridges at two binding sites: E27 from CXCR1 and R47 from IL8 in site I (Figs.
3.4. Stability and binding modes of CXCR4-vMIP-II with and without N terminal residues
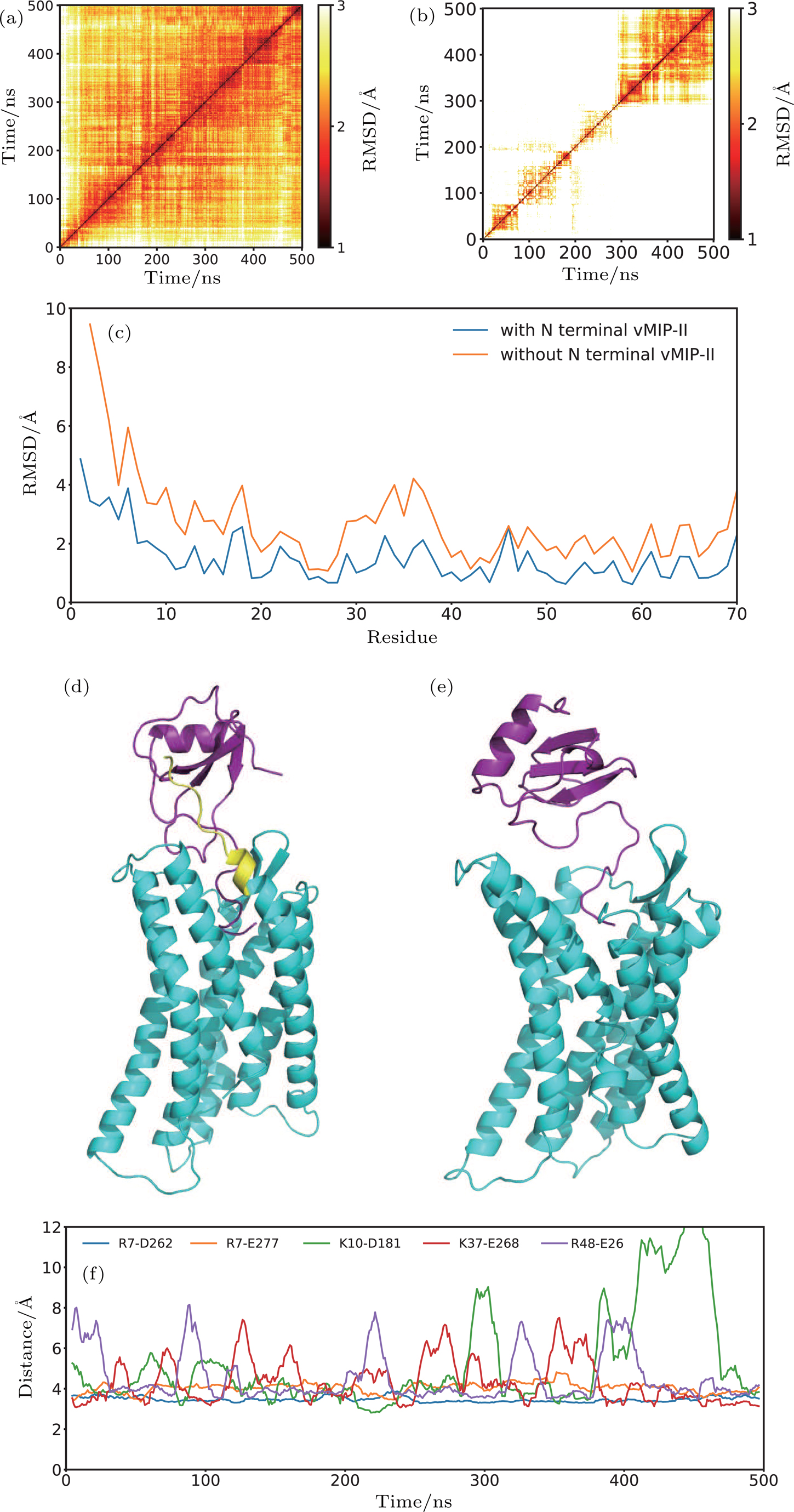
The comparative study of CXCR1-IL8 systems suggested the critical roles of N-terminal of the receptor in stabilizing the ligand binding. We carried out the simulations to the crystallography model of CXCR4-vMIP-II complex as a control study. The 11 residues at N-terminal (S24-N34) of the CXCR4 were truncated in the modified system (see Fig.
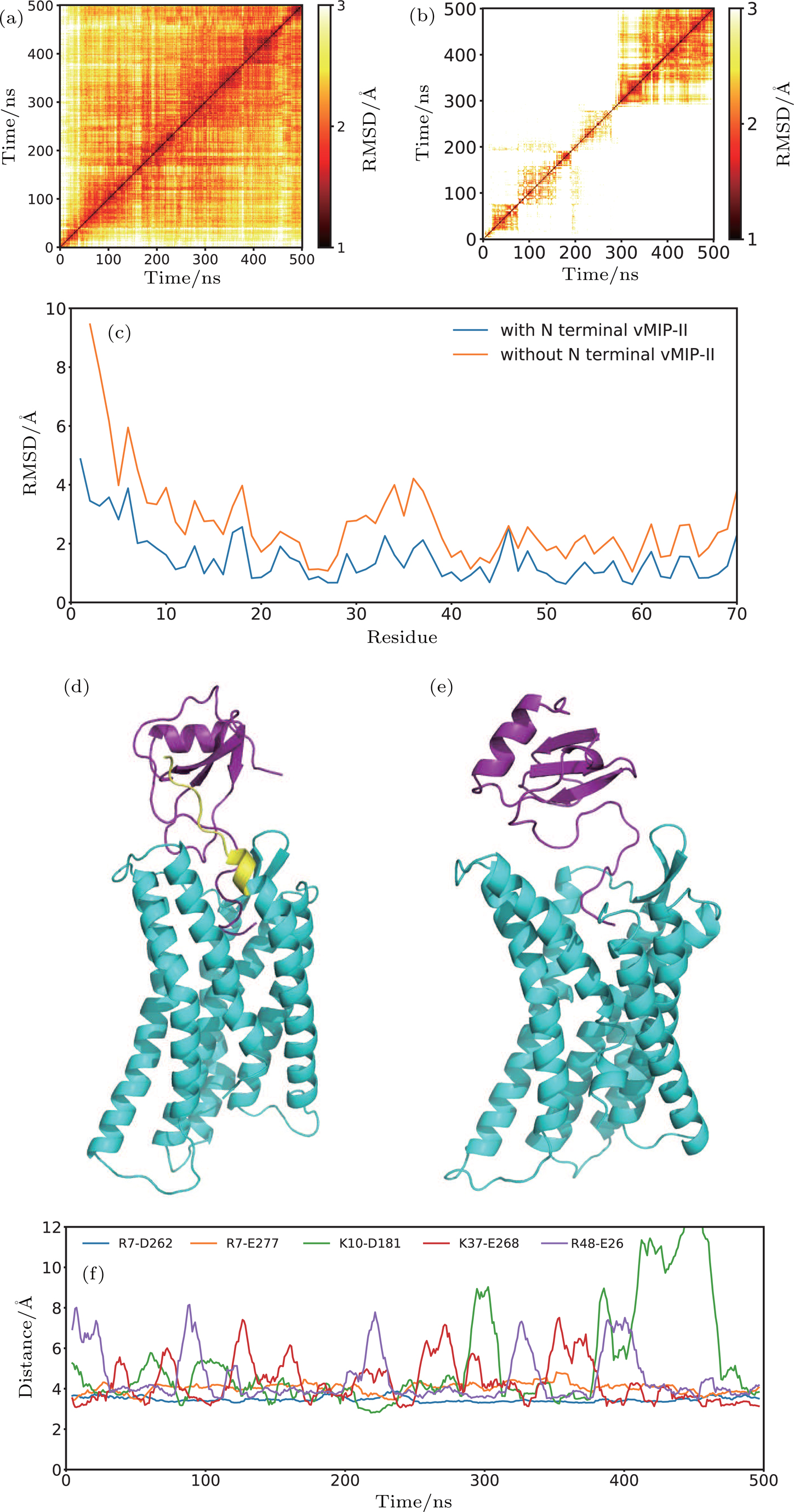 | Fig. 7. (color online) The structure and dynamics of CXCR4-vMIP-II complex. (a), (b) 2D RMSD of CXCR4-vMIP-II complex with N terminal (a) and without N terminal (b). (c) The RMSF of vMIP-II for CXCR4-vMIP-II complex with N terminal (blue) and without N terminal (orange). Final structures of CXCR4-vMIP-II complex with N terminal (d) and without N terminal (e). (f) Distance between all the salt bridge pairs at the interface of the CXCR4-vMIP-II complex structure (PDB ID 4RWS). The distance curve was smooth by the window of 10 ns. |
4. Discussion and conclusion
In this work, we present the results on GPCR protein in a complex with a ligand, the CXCR1-IL8, using the homology modeling and all atom molecular dynamics simulation methods. The data shows that the predicted models are plausible, providing a binding pose stabilized by electrostatic interactions at two major binding sites. The comparative study of different models also suggests that the N-terminal of CXCR1 plays critical roles in establishing the favorable interactions at binding site I, reflected by the fact that the shorter N-terminal in WT model 2 leads to unstable ligand binding. In order to better understand the results, we would like to discuss the issues related to the results presented in the paper.
4.1. Validity of homology models and the plausibility of the binding mode
For template-based structure prediction, the well-aligned region can have high accuracy in the structure prediction, if the sequence similarity is above 30%.[40,41] The CXCR1 and CXCR4 have high sequence similarity (about 38% for full sequence similarity and about 42% for TM region similarity) in our case, permitting a good sequence alignment for 3D structure prediction. Furthermore, the structure of CXCR1 without ligand was resolved using the MR method, providing a cross validation model for the predicted model based on CXCR4. The ligand molecules vMIP-II and IL8 are also similar in both sequence (about 21% for sequence similarity) and structure (RMSD value is 1.9 Å between v-MIP-II and IL8). Therefore, the predicted complex structures are very plausible.
The simulation data for WT model 2, which is based on CXCR1 structure determined using NMR method, showed an unstable complex. It could be due to two reasons: (i) the interactions at the binding site are lost due to the missing N-terminal residues in CXCR1 structure; and (ii) the binding pocket of the NMR model is small because the structure does not contain a ligand molecule, therefore the CXCR1 structure has a ‘closed’ binding site II. In a previous study, the computational docking method was applied to predict the binding poses of IL8 to CXCR1, without the information of CXCR4-vMIP-II complex. In the predicted models, the N-terminal of IL8 was never observed inside the binding pocket.[29] This could be due to the artifacts of the docking protocols, because docking to the deep pocket is often challenging, as in the case of the binding site II in CXCR1.
4.2. The roles of receptor protein N-terminal
The roles of the N-terminal are also investigated using a homology system, the CXCR4-vMIP-II complex. The removal of N-terminal residues from the complex structure leads to unstable binding of vMIP-II to the CXCR4, supporting the discovery in the CXCR1-IL8 system.
Previous work has suggested that the receptor–ligand binding reaction between IL8 and CXCR1 is a multistep process: started by weak and nonspecific complex formation between the N terminal of CXCR1 and IL8, followed by subsequent cooperative rearrangement between the ligand and the receptor, and finally obtained a more compact and specific final structure.[42] Although modeled from different templates, the final structures of WT model 1 and extended model show similar binding position and possess consistent features at two binding sites. The N terminal of CXCR1 plays important roles in stabilizing the ligand binding by forming salt bridges between CXCR1 and IL8 at binding site I. Changing the N terminal sequence of CXCR1 to CXCR4 reduces the stability and the number of salt bridges between the IL8 and CXCR1. Nonetheless, the binding position could be maintained because of the binding site I, as shown in the case of the hybrid model. The removal of the N-terminal of CXCR1 in the structure leads to the unstable complex structure, although the individual ligand or receptor remains stable through the simulations. Without the N-terminal residues of CXCR1, the IL8 has larger freedom, reflected in the rotation around the Z-axis and the tendency of dissociation from the CXCR1 complex, as observed in the WT model 2. The previous structural work of IL8 with N terminal fragment of CXCR1 shows evidence of well-defined electrostatic interactions between K11, K15, R47 of IL8 and D24, E25, D26 of CXCR1 N-terminal residues, which were observed in the WT model 1.[24,25] On the other hand, these stable salt bridges found in binding site II are consistent with the previous mutagenesis studies, which demonstrated that the mutations of E275A, D288A, and E291A lower the binding affinity of CXCR1 to IL8.[26] Those experimental data suggested that the charged residues near ECL3 were crucial and the interaction between R6 from IL8 and E275 from CXCR1 were important for the binding. The relative weaker interaction in the hybrid model might be due to the difference in stable salt bridges (see Table
4.3. The prospect of simulation results for experimental structure determination
Given the model and stability results obtained from extensive simulations, we propose that the CXCR1-IL8 complex structure is stabilized by the interactions at two binding sites. The binding mode for different models (except the WT model 2) is similar after 500 ns simulations. This provides a good starting model to guide the protein engineering to design constructs that have improved stability. For example, based on the predicted structures, disulfide bonds between CXCR1 and IL8 can be introduced to improve the thermal stability of the complex. This has been demonstrated in a recent work by Liu and coworkers, who developed a computational algorithm to predict possible cysteine mutation sites to form disulfide bonds based on given structures that can be from crystallography or homology modeling.[43]
In this work, several models with sequence difference are compared. Although the results still need further experimental validation, the conclusion is sound and consistent. We believe that structure prediction combined with all-atom MD simulation can be used as a general method to investigate the structure and dynamics, even before the structures are experimentally determined. Nonetheless, the correctness of the starting model is crucial, i.e., the homology modeling should be at high confidence levels before carrying out large-scale, all-atom, molecular dynamics simulations. Given that more diverse protein structures are available in the database, the homology modeling accuracy will be continuously improved. We predict that the molecular dynamics simulation will become applicable for many other molecular systems, and be helpful for the experimental structure determination.
In summary, the structure of CXCR1-IL8 complex is predicted using the homology modeling method and the stability is further studied using molecular dynamics simulations. Key residues that form salt bridges at the CXCR1-IL8 interfaces are also identified. The information obtained in this study can be applied in designing engineered proteins to improve complex stability.
Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] | |
| [33] | |
| [34] | |
| [35] | |
| [36] | |
| [37] | |
| [38] | |
| [39] | |
| [40] | |
| [41] | |
| [42] | |
| [43] |