



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Computational study of non-catalytic T-loop pocket on CDK proteins for drug development
Cite this Article
Wang Huiwen, Wang Kaili, Guan Zeyu, Jian Yiren, Jia Ya, Kashanchi Fatah, Zeng Chen, Zhao Yunjie. Computational study of non-catalytic T-loop pocket on CDK proteins for drug development. Chinese Physics B, 2017, 26(12): 128702 
Permissions
Computational study of non-catalytic T-loop pocket on CDK proteins for drug development
† Corresponding author. E-mail:
Abstract
Cyclin-dependent kinases (CDKs) are critical to the cell cycle and many other biological processes, and as such, are considered as one of the promising targets for therapy against cancer and other diseases. Most pan-CDK inhibitors bind to the highly conserved catalytic ATP-binding pocket and therefore lack the specificity to prevent side effects. It is desirable to develop drugs targeting non-catalytic pockets for specificity towards individual CDKs. Here we performed a systematic analysis of non-catalytic pockets on CDKs and identified a region underneath the T-loop, which we term TL pocket, for potential inhibitor development. Specifically, we compared the TL pockets of human CDK2 and CDK7-homolog Pfmrk of Plasmodium falciparum, a malaria-causing parasite. Molecular dynamics simulations of several short peptides revealed that this less conserved TL pocket could be used to design potentially specific inhibitors against malaria disease.
1. Introduction
Cyclin-dependent kinases (CDKs) are a family of protein kinases that regulate the cell cycle and many other biological processes.[1] For example, CDK1, 2, 4, and 6 are directly involved in regulating the cell cycle; CDK5 is required for proper development of the brain; CDK7, 8, and 9 are part of the elongation factor for RNA polymerase II transcription.[2–7] Structurally, CDK proteins consist of an N-terminal lobe rich in beta strands, C-helix, alpha helical, and C-terminal lobe. ATP binds to CDKs in a deep cleft between the two lobes. Cyclin binding to CDKs tightly regulates their functions. Aberrant activity of various cell cycle proteins can result in uncontrolled tumor cell proliferation. Therefore, CDK proteins are considered attractive drug targets.[8–12] For example, current therapy against malaria becomes increasingly less effective due to drug resistance in the malaria-causing parasite; it is thus highly desirable to develop new drugs targeting the parasite’s CDK required for its life cycle.[13–18]
CDK activation is a two-step process. In the inactive state, T-loop arises from the C-terminal lobe to block the binding of a protein substrate at the entrance of the active site cleft. So, the first step is the binding of the regulatory protein Cyclin to decrease the T-loop flexibility. Second, the Cyclin protein can also position several essential amino-acid side chains correctly, so the phosphates of ATP can be ideally oriented for the kinase reaction. Therefore, previous CDK drug development focused on blocking ATP or breaking up the CDK/Cyclin interface.
Currently, most of the CDK inhibitors are ATP-competitive. Although with some slight differences, the catalytic ATP-binding pockets of CDK family proteins share a similar structure, the pan-CDK inhibitors typically exhibited little specificity towards individual CDKs. For example, flavopiridol is a well-known pan-CDK inhibitor and can inhibit CDK1, 2, 4, and 9.[19–22] Previous study and clinical trials indicated that flavopiridol causes the cell cycle arrest in G1 and G2 phases but also lead to tissue apoptosis or organ atrophy. Another approach for drug design is to break up the CDK/Cyclin complex. However, it is noted that the buried surface at the interface of CDK/Cyclin is extensive with hydrophobic interactions. It is difficult to design a small compound to compete directly with or break up Cyclin protein at the interface. However, recent computational studies suggest that it is possible to design small compounds to break up Cyclin protein at non-catalytic pocket via allosteric interactions.[23–29] Stephane et al. identified one non-catalytic pocket away from the ATP site that extends from the DFG region above the C-helix.[30] Crystal structures with ANS revealed that two ANS molecules were bound adjacent to each other. Binding of ANS induced some structural changes in the C-helix conformation that made Cyclin binding impossible. Giulio et al. further developed the allosteric inhibitors of cyclin-dependent kinase 2 using this non-catalytic pocket.[31] Previously, we reported some small peptides targeting a non-catalytic TL pocket located under the T-loop of CDK protein to break up the CDK2/Cyclin complex.[32] Computational dynamical network analysis revealed that these peptides weaken the complex via allosteric interactions. Our experiments showed that upon binding to the non-catalytic pocket, these peptides break up the CDK2/Cyclin complex partially and diminish its kinase activity in vitro. Yutong et al. further developed the allosteric chemical small-molecule CDK2 inhibitor based on this non-catalytic pocket.[33] These results indicate that the non-catalytic pocket may be utilized as a potential drug target for other CDK proteins. However, the specificity of the non-catalytic pocket towards individual CDKs remains poorly understood.
In this paper, we present a systematic analysis of CDKs to identify all potential non-catalytic pockets. Results on sequence evolution suggest that the non-catalytic TL pocket located under the T-loop is much less conserved in both sequence and structure than the ATP-binding pocket. The case study of the malaria-causing parasite kinase Pfmrk, a sequence homolog of human CDK7, shows that the interface regions of Pfmrk/Cyclin H complex would be weakened differently from that of human CDK2/Cyclin E by certain peptide inhibitors. Taken together, we provide a scheme to identify non-catalytic pockets and further demonstrate that the non-catalytic TL pocket could be used to develop drugs with specificity towards the parasite and reduced side effects on host cells.
2. Materials and methods
2.1. Pocket detection
Potential binding cavities were detected using active site identification program DoGSiteScorer.[34,35] The program identifies all cavities on the surface of a given protein structure and then calculates the global properties of the cavity including its volume, surface, shape and chemical features (see Refs. [34] and [35] for details). DoGSiteScorer[34,35] has correctly classified druggable or undruggable cavities with an accuracy of 90% in a non-redundant data set (NRDD) and accuracy of 88% in a druggability data set (DD). Therefore, this program could reliably discover potential cavities for drug binding. PyMOL was used to visualize the protein structures and their putative cavities (
2.2. Conservation analysis
The CDK2, CDK7, and CDK9 structures were extracted from the PDB database (PDB codes: 1FIN, 1UA2, and 3MI9, respectively).[39–41] The parasite CDK Pfmrk and human Cyclin H complex was built via homology modeling by I-TASSER.[42,43]
The homologous sequences of CDK2, CDK7, and CDK9 proteins were extracted from ConSurf-DB.[44,45] The CSI-BLAST is used to search for the homologous sequences similar to the selected structure. Then, the multiple sequence alignment (MSA) of these homologous sequences is calculated by MAFFT.[46] The position-specific conservation scores of each amino acid position in the alignment were computed using the Rate4Site program.[47] This Rate4Site algorithm assigns a conservation level for each residue using an empirical Bayesian inference. Visualization of the conservation patterns on tertiary structure often provides insights on functionally important regions of the protein. Tertiary structural conservation patterns were visualized using PyMOL (
2.3. Molecular dynamics simulation and inhibitor evaluation
All molecular dynamics (MD) simulations were performed using the GROMACS software package.[48] We have done a benchmark simulation test in our previous CDK2 study with experimental validation.[32] Here we used the tested MD parameters in our current Pfmrk MD study. The peptide inhibitor structures were exactly the same as described in our previous work.[32] For the MD simulations, the G53a6 force field and SPC water were employed.[49] The temperature was set at 300 K. Before MD simulations, the entire systems were first minimized by a 1000-step steepest descent calculation followed by a 3000-step conjugate gradient optimization. We performed 30-ns MD simulations for each different state. The dynamical protein network is constructed as follows. A node is defined as a single amino acid, and if the distance of any two heavy atoms of a pair of different nodes is less than 4.5 Å for at least 75% of all snapshots sampled during MD simulations, then this pair of nodes was said to form an edge.[50,51] The neighboring nodes in the sequence were not considered to be in contact and thus no edge between them. The final 20 ns of the 30-ns trajectories, sampled every 100 ps, was used to construct the protein network. Moreover, we define the pairwise correlations
3. Results
3.1. Potential drug pockets on CDK proteins
Accurate identification of pockets is important in the study of computational drug design, therefore, we identified all the potential drug binding pockets of CDK2, CDK7, and CDK9 using DoGSiteScorer program.[34,35] There are putatively 13, 9, and 15 pockets on CDK2, CDK7, and CDK9 structural surface, respectively. The approximate volumes and surfaces of different pockets are given in Tables
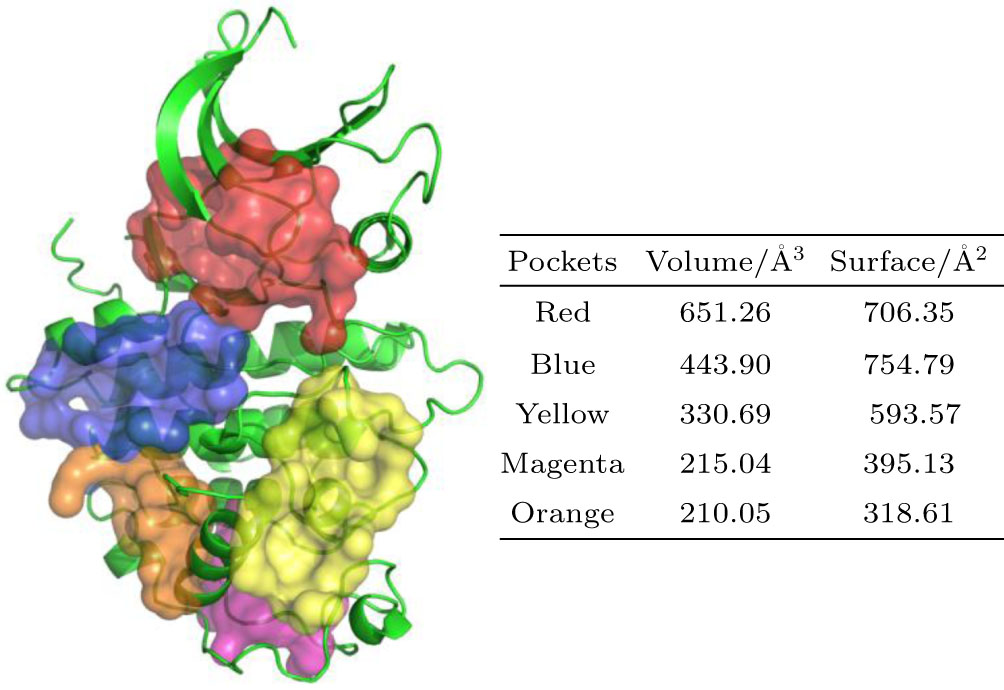 | Fig. 1. (color online) The top five detected pockets (shown as surface representation) together with a cartoon representation of CDK2 protein (PDB code: 1FIN). The main shape descriptors (volume and surface) for the top 5 detected pockets are shown in the table on right. |
| Table 1. The shape descriptors (volume and surface) for all detected pockets on CDK2 structural surface (PDB code: 1FIN). All the pockets are identified using DoGSiteScorer[34,35] program. . |
3.2. Conservation analysis of pockets
Given that protein structures evolve,[53] we performed sequence conservation analysis to infer the structural or functional important residues. The evolutionary conservation scores were identified using the ConSurf-DB.[44,45] The continuous conservation scores are divided into a discrete scale of 9 grades with grade 1 indicating the most variable positions and grade 9 the most conserved positions. The highly conserved residues (grades 7–9), variable residues (grades 1–3), and pocket conservations of CDK proteins are listed in Tables
| Table 4. The conservation analysis of CDK2 structure (PDB code: 1FIN). The evolutionary conservation scores are identified using the ConSurf-DB.[44,45] The continuous conservation scores are divided into a discrete scale of 9 grades with grade 1 for the most variable positions while grade 9 the most conserved positions. . |
| Table 5. The conservation analysis of CDK7 structure (PDB code: 1UA2). The evolutionary conservation scores are identified using the ConSurf-DB.[44,45] The continuous conservation scores are divided into a discrete scale of 9 grades with grade 1 for the most variable positions while grade 9 the most conserved positions. . |
| Table 6. The conservation analysis of CDK9 structure (PDB code: 3MI9). The evolutionary conservation scores are identified using the ConSurf-DB.[44,45] The continuous conservation scores are divided into a discrete scale of 9 grades with grade 1 for the most variable positions while grade 9 the most conserved positions. . |
| Table 7. The pocket conservation score of CDK2 structure (PDB code: 1FIN). The first and second columns represent the average conservation score of the pocket and all residues in the pocket. . |
| Table 8. The pocket conservation of CDK7 structure (PDB code: 1UA2). The first and second columns represent the average conservation score of the pocket and all residues in the pocket. . |
| Table 9. The pocket conservation of CDK9 structure (PDB code: 3MI9). The first and second columns represent the average conservation score of the pocket and all residues in the pocket. . |
We have projected the evolutionary conservation scores of each amino acid onto the CDK tertiary structures (shown in Fig.
 | Fig. 2. (color online) Ribbon and surface representation of (a) CDK2 (PDB code: 1FIN), (b) CDK7 (PDB code: 1UA2), and (c) CDK9 (PDB code: 3MI9). Color scheme follows the conservation scores. Variable residues (conservation score from 1 to 3), average residues (conservation score from 4 to 6), and conserved residues (conservation score from 7 to 9) are colored in blue, green and red, respectively. Both the ATP-binding pocket and protein structural core are highly conserved as marked in red. The non-catalytic T-Loop pocket (TL pocket) has more sequence and structural variations. |
Previously, we reported some short peptide inhibitors targeting this non-catalytic TL pocket.[32] Molecular dynamics simulations and detailed dynamical network analysis revealed that these peptides weaken the complex formation via allosteric interactions. Our experiments also showed that upon binding to the non-catalytic TL pocket, these peptides break the CDK2/Cyclin complex partially and diminish its kinase activity in vitro. Given that this non-catalytic TL pocket is less conserved, the variation might be exploited to design specific drugs for different CDK proteins.
3.3. Specific peptide inhibitors for the non-catalytic TL pocket on Pfmrk for potential malaria therapy
We can explore the variability of non-catalytic binding pockets of CDKs to not only reduce cross interactions for inhibitors against different CDKs of the same organism but also potentially design inhibitors against a specific parasitic organism while leaving the host organism intact. To this end, we focus on CDK7-homolog Pfmrk from Plasmodium falciparum, a malaria-causing parasite that kills a significant number of lives in the world, especially in the developing countries.[54] Given the emergence and spread of drug-resistant malaria, new drugs against malaria are urgently needed. Pfmrk is an attractive drug target due to its role in regulating the parasite’s cellular proliferation. It was reported that Pfmrk forms a stable complex with human Cyclin H and stimulates kinase activity.[55] We thus aim to design inhibitors targeting the non-catalytic TL pocket that can break Pfmrk/Cyclin H complex while leaving CDK2/Cyclin E of the host human cell unaffected.
To visualize the variability of the ATP-binding pocket and the non-catalytic TL pocket between human and parasite Plasmodium, we constructed the phylogenetic tree of these two pockets as shown in Fig.
 | Fig. 3. (color online) Phylogenetic tree of the ATP-binding pocket (a) and the non-catalytic T-loop (TL) pocket (b). Human CDK2 and parasite CDK Pfmrk are highlighted in red and blue. The non-catalytic TL pocket is much less conserved in both sequence and structure than ATP-binding pocket. |
It is known that Cyclin binding to CDK modulates the enzymatic kinase activity of CDK. A stable interface of CDK/Cyclin is thus required. Weakening the interface could be a strategy for inhibitor design. Indeed, it was the approach used in our previous studies[32] where MD simulations were performed to monitor the interface motion caused by peptide inhibitors placed in the TL pocket. The interface stability is measured by the interface correlation via dynamical network analysis (see Methods for details). Larger interface correlation indicates stronger interface coupling and thus more stable complex. As shown on the top panel of Fig.
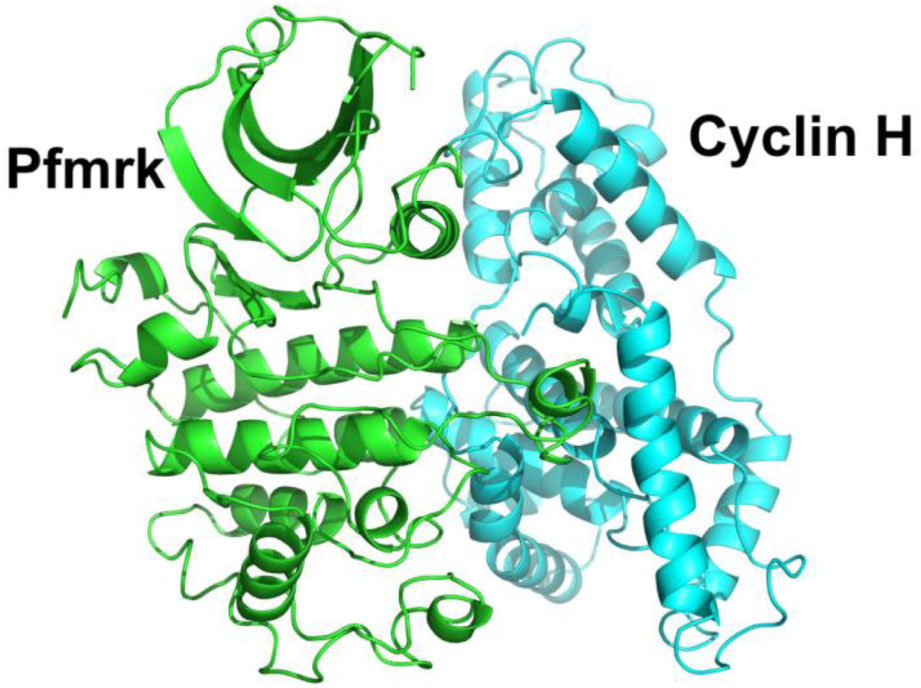
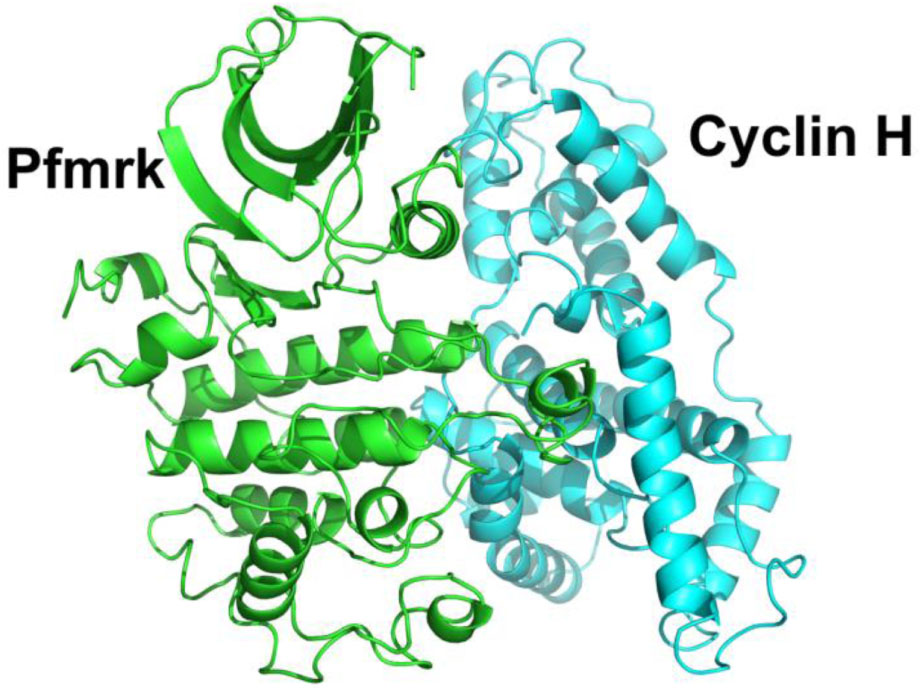 | Fig. 4. (color online) Ribbon representation of the parasite CDK Pfmrk and human Cyclin H complex. The structure was built via homology modeling by I-TASSER using human CDK7 and Cyclin H complex. Pfmrk is colored in green. Cyclin H is colored in light blue. |
 | Fig. 5. (color online) Correlation strength of CDK/Cyclin interface with and without 5mer peptides located at the TL pocket of CDK2/Cyclin E (a) and Pfmark/Cyclin H (b). The correlation was computed by the dynamical network analysis of MD simulations. The most decreased interface correlations were produced by DAALT and YAALQ on CDK2/Cyclin but FAALA and RAALW on Pfmrk/Cyclin showing specificity to some degree. The computational results for CDK2/Cyclin E are consistent with previous experiments (refer to the main text). |
Previous research showed that highly coupled residues with common secondary structure elements will produce a correlation value great than 0.7, while some other secondary structure elements with strong interactions can also achieve correlation value around 0.5 ∼ 0.6. Residues across an interface typically result in correlations ranging from 0.3 to 0.4.[32] With the experimental validation of our computational studies on the interface stability of human CDK2/Cyclin E complex, which showed that a decreased interface correlation corresponds to weakened interface stability,[32] we further probed how these same peptide inhibitors might impact the interface stability of Plasmodium Pfmrk/Cyclin H complex. To this end, we first built the Pfmrk/Cyclin H complex via homology modeling as shown in Fig.
4. Discussion
Cross-interaction due to the lack of specificity is a common problem in drug design. Targeting less-conserved non-catalytic residues offers a further opportunity to design drugs with desired specificity to reduce the risk of side effects. This approach could be applied to other kinase proteins. Some studies have extended this approach to non-kinase proteins. For example, Hagel et al. developed non-catalytic initiators for Hepatitis C virus (HCV) protease.[56] In this study, we performed a systematic analysis of CDK sequences and identified all potential binding pockets with a particular focus on the non-catalytic TL pocket and its potential for designing a new class of inhibitors distinct from the traditional APT-competitive inhibitors. We demonstrated, as an example, that this variable TL pocket could indeed be explored to design specific peptide inhibitors against kinase Pfmrk from malaria-causing parasite Plasmodium falciparum with minimal impact on human CDKs of the host cell. While our predictions on Pfmrk are only computational so far, which must be further verified experimentally, the results are tantalizing enough to merit further optimization on the peptide inhibitors based on the subtle difference of the TL pocket of CDKs among different organisms.
In summary, focusing only on the CDK branch of an entire kinome for different organisms, we provided a computational approach that combines structure modeling, pocket detection, and evolutionary conservation analysis to identify non-catalytic pockets for designing inhibitors of the desired specificity. It could be useful to extend this approach to the entire kinome.
Author contributions
Huiwen Wang and Kaili Wang performed most computational analysis under the supervision of Ya Jia. Fatah Kashanchi helped with the research design; Yunjie Zhao performed MD simulations; Zeyu Guan and Yiren Jian helped with the conservation analysis; Yunjie Zhao and Chen Zeng supervised the overall study and wrote the paper.
Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] | |
| [33] | |
| [34] | |
| [35] | |
| [36] | |
| [37] | |
| [38] | |
| [39] | |
| [40] | |
| [41] | |
| [42] | |
| [43] | |
| [44] | |
| [45] | |
| [46] | |
| [47] | |
| [48] | |
| [49] | |
| [50] | |
| [51] | |
| [52] | |
| [53] | |
| [54] | |
| [55] | |
| [56] |