


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Passivation effects of phosphorus on 4H-SiC (0001) Si dangling bonds: A first-principles study
Cite this Article
Li Wenbo, Li Ling, Wang Fangfang, Zheng Liu, Xia Jinghua, Qin Fuwen, Wang Xiaolin, Li Yongping, Liu Rui, Wang Dejun, Pan Yan, Yang Fei. Passivation effects of phosphorus on 4H-SiC (0001) Si dangling bonds: A first-principles study. Chinese Physics B, 2017, 26(3): 037104 
Permissions
Passivation effects of phosphorus on 4H-SiC (0001) Si dangling bonds: A first-principles study
† Corresponding author. E-mail:
Abstract
The effect of phosphorus passivation on 4H-SiC(0001) silicon (Si) dangling bonds is investigated using ab initio atomistic thermodynamic calculations. Phosphorus passivation commences with chemisorption of phosphorus atoms at high-symmetry coordinated sites. To determine the most stable structure during the passivation process of phosphorus, a surface phase diagram of phosphorus adsorption on SiC (0001) surface is constructed over a coverage range of 1/9–1 monolayer (ML). The calculated results indicate that the 1/3 ML configuration is most energetically favorable in a reasonable environment. At this coverage, the total electron density of states demonstrates that phosphorus may effectively reduce the interface state density near the conduction band by removing 4H-SiC (0001) Si dangling bonds. It provides an atomic level insight into how phosphorus is able to reduce the near interface traps.
1. Introduction
Silicon carbide (SiC) has a wide application in extremely harsh environments. Besides, it is also a promising wide-band-gap semiconductor material for fabrication of high-temperature, high-frequency, and high-power metal–oxide–semiconductor (MOS) devices due to its excellent physical properties as well as native oxide (SiO2).[1–4] Among the commercially available SiC polytypes, 4H-SiC is most attractive for high-power electronic devices such as MOS field effect transistors (MOSFETs) owing to its higher bulk electron mobility and smaller anisotropy.[5,6] However, the high density of interface traps (Dit) near the conduction band edge at the SiO2/SiC (0001) interface gives rise to the low channel mobility of 4H-SiC MOSFETs.[7–10] It is therefore important to passivate the interface to improve the channel mobility of SiC MOSFETs.
A large number of process methods have been tried to reduce the defect density to an acceptable level. It has been demonstrated experimentally that the interface state densities can be significantly reduced by annealing SiO2/SiC interfaces with nitrogen (N).[11–21] Unfortunately, the obtained Dit (∼ 2 × 1011 cm−2·eV−1) after nitridation treatment is still too high for SiC MOS capacitors.[13] It is necessary to explore new passivation processes to achieve higher mobility for 4H-SiC MOSFETs. Okamoto et al. recently attempted to reduce the Dit by post-oxidation annealing in phosphoryl chloride (POCl3).[22–25] Their results showed that the Dit was effectively reduced by phosphorus (P) atoms and the peak field-effect mobility of 4H-SiC MOSFETs could be increased to 89 cm2/V·s. Their subsequent studies suggested that the positive effects of P atoms might be attributed to the passivation of NITs, in particular, the elimination of Si–Si bonds.[26] However, atomic scale understanding of phosphorus at the interface has remained unclear.
Experiments have reported many kinds of defects exist at the SiO2/SiC interface. The dangling bond defect, as a simple interface defect, is also found at the SiO2/SiC interface,[27] similar to the SiO2/Si interface.[28] Pennington and Ashman recently used density functional theory (DFT) calculations to investigate nitrogen passivation of 4H-SiC (0001) Si-face dangling bonds.[19–21] That work showed that the Si dangling bonds may create states below the conduction band edge and the dangling bond defect states may be removed by nitrogen. However, little is still known about the effects of phosphorus passivation on the dangling bonds. In this work, we study the effect of phosphorus passivation 4H-SiC (0001) Si dangling bonds by using the first-principles approach, in order to better understand the removal mechanism of the defect states.
2. Computation methods
where NP is the number of P atoms adsorbed on the 4H-SiC (0001) surface, and EP, Eslab, and EP/slab refer to the total energies of the isolated P atom, and the surfaces with and without the P adatoms, respectively.
where µP is the chemical potential of P atom. Combining Eqs. (1 ) and (2 ), one can obtain
with ΔµP = µP − EP.
DFT calculation is performed using Cambridge serial total energy package (CASTEP) code. The generalized gradient approximation (GGA) function proposed by Perdew and Wang (PW91)[29] and a plane-wave cutoff of 300 eV are employed after a full convergence test. The Vanderbilt ultrasoft pseudopotentials[30] are used to describe the ion–electron interaction. The equilibrium atomic positions are determined when the maximum Hellmann–Feynman force and the change of the total energy are below 0.03 eV/Å and 0.01 meV, respectively. The optimized lattice constants are a = 3.078 Å and c = 10.046 Å using a 9 × 9 × 2 Monkhorst–Pack grid, agreeing well with the experimental values (a = 3.073 Å and c = 10.053 Å).
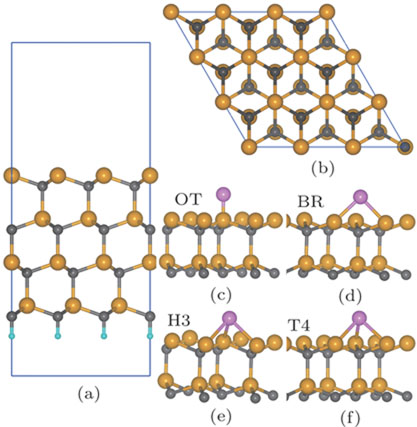
The 3× 3 × 1 4H-SiC (0001) supercell consists of four bilayers of SiC with a vacuum space of 10 Å in dimension. The carbon dangling bonds at the bottom layer are terminated by hydrogen atoms and the bottom three layers are fixed to simulate the infinitely large bulk solid. A converged 3 × 3 × 1 Monkhorst–Pack k-point mesh[31] is used during the geometry optimizations of various surface models. On the SiC (0001) surface, P adatoms are initially placed on four high-symmetric adsorption sites as shown in Fig.
 | (1) |
 | Fig. 1. (color online) (a) Side and (b) top views of 4H-SiC (0001) surface and (c), (d) four adsorption sites on this surface. The on-top site (OT) is located on the top of a surface Si atom, the bridge site (BR) is a two-fold coordinated site between two surface Si atoms, the three-fold hollow site (H3) is above a carbon of the third bilayer, and the four-fold hollow site (T4) is above a carbon of the first bilayer. The orange, grey, magenta, and cyan spheres denote silicon, carbon, phosphorus, and hydrogen atoms, respectively. |
The stability of each structure is assessed by calculating the Gibbs free energy of formation ΔGform in the grand canonical ensemble
 | (2) |
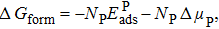 | (3) |
3. Results and discussion
We start our analysis by studying P atom adsorption on the 4H-SiC (0001) surface. For a single P atom adsorption, corresponding to 1/9 monolayer (ML) coverage in our supercell model, only the H3 and T4 binding configurations are stable. According to the calculated result, the H3 site is more favorable since its adsorption energy is 0.19 eV higher than that of the configuration with a P atom at the T4 site. For the 2/9 ML coverage surface, five possible stable configurations are obtained after optimizing 37 initial structures. Unlike the nitrogen adsorption on 4H-SiC,[18] the most energetically favorable configuration is the one with two P atoms at different T4 sites rather than different H3 sites. It is interesting that the most stable location for P atoms shifts from the H3 site to T4 site. This might be attributed to the strong interaction between two P atoms which makes the P adatoms deviate from the H3 sites. When three P atoms are deposited on the supercell of 4H-SiC surface (i.e., 1/3 ML coverage), these P atoms still prefer separated T4 sites. It is also found that this coverage has a 
The dependence of the adsorption energy per P atom on the phosphorus coverage is illustrated in Fig.
 | Fig. 2. The Gibbs free energy of formation as a function of phosphorus coverage. |
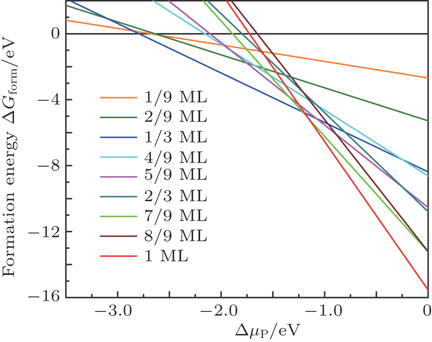 | Fig. 3. (color online) Adsorption energy per phosphorus adatom as a function of phosphorus chemical potential. |
To determine the stability of various P/slab configurations, we plot the Gibbs free energy of formation as a function of phosphorus chemical potential using Eq. (
Figure
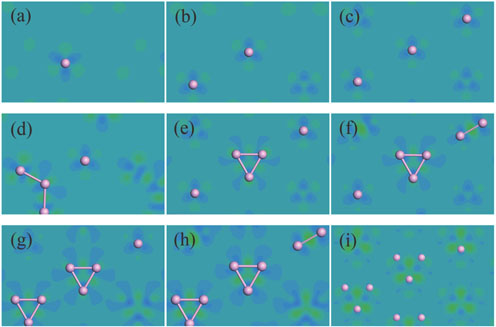
 | Fig. 4. (color online) Different electron densities of the stable configurations for the coverages of (a) 1/9 ML, (b) 2/9 ML, (c) 1/3 ML, (d) 4/9 ML, (e) 5/9 ML, (f) 2/3 ML, (g) 7/9 ML, (h) 8/9 ML, (i) 1 ML. The violet spheres denote phosphorus atoms. |
To further elucidate the phosphorus passivation effects on the density of interface traps, we present the electron densities of states (DOS) for the clean surface, 1/9 ML, 1/3 ML, and 4/9 ML coverage as shown in Fig.
 | Fig. 5. (color online) Total densities of states for the clean surface and surfaces with 1/9 ML, 1/3 ML, and 4/9 ML phosphorus coverages. |
4. Conclusion
We investigate adsorption behaviors of P atoms on 4HSiC (0001) and effects of P passivation on surface Si dangling bonds by using density functional theory methods. The surface diagram result indicates that the 1/3 ML configuration is most energetically favorable (i.e., a reasonable phosphorus concentration). At this coverage, the density of surface states within the band gap of bare surface is found to be decreased. It also shows that P passivation removes states from the conduction band gap. This is similar to the previous experimental observation[22,26] even though a complicated oxide during P passivation is not theoretically considered. Therefore, the physical mechanisms of P passivation of the bare SiC surface may also play a role during P passivation of the SiO2/SiC interface. The presence of P atoms at the SiO2/SiC interface region can effectively eliminate Si dangling bonds and decrease the interface state density.
Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] |