{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Modulating doping and interface magnetism of epitaxial graphene on SiC(0001)
[Zhou Pan , He Da-Wei †,  ]
]
]
|
|


† Corresponding author. E-mail:
Project supported by the National Natural Science Foundation of China (Grant Nos. 61335006, 61378073, and 61527817), the Beijing Municipal Science and Technology Committee, China (Grant No. Z151100003315006), and Fundamental Research Funds for the Central Universities of Beijing Jiaotong University, China (Grant No. 2012YJS123).
On the basis of first principles calculations, we report that the type and density of charge carriers of epitaxial graphene on SiC(0001) can be deliberately controlled by decorating the buffer layer with specific atoms (i.e., F, Cl, O, or N). More importantly, a fine tuning of the doping behavior from intrinsic n-type to charge neutrality to p-type and interface magnetism is achieved via increasing the doping concentration of F atoms on the buffer layer. Our results suggest an interesting avenue to the application of epitaxial graphene in nanoscale electronic and spintronic devices.
The distinct electronic structure and physical properties of graphene [ 1 – 4 ] or graphene-like two-dimensional materials [ 5 ] have aroused considerable interest. More particularly, graphene has been made a candidate of Si in the post-Moore era. [ 6 – 9 ] Nevertheless, the successful development of graphene-based electronics requires the large-scale synthesis of high-quality graphene. The epitaxial growth of graphene on SiC(0001) or 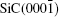
On the other hand, interface engineering has been widely used to modify the structural and electronic properties in the heterojunctions, such as BNC system, [ 31 ] carbon nanotubes on InAs(111), [ 32 ] Si(0001) [ 33 ] surface, epitaxial graphene on Ir(111). [ 34 ] Wang et al. proposed that n-doped or p-doped graphene sheets on a chip could be achieved by modifying the SiO 2 substrate with self-assembled layers of silane and polymer in experiment. [ 35 ] Indeed, in the system of epitaxial graphene on SiC, Zhou et al. experimentally showed that the doping of graphene was associated with the surface charges at the interface, rather than the carrier concentration of the substrate. [ 36 ] Thus the first graphene layer and surface of the substrate, acting as the interface, offer the possibility of using them for band engineering of the system. [ 37 , 38 ] Some studies introduced a configuration with graphene/intercalated atoms/substrate for investigating the decoupling of graphene and substrates by other atoms. [ 10 , 34 , 39 ] In the system of epitaxial graphene on Si-terminated substrate, the first graphene layer and the substrate are linked by covalent bonds, which need high energy consumption to be broken up. Meanwhile, the first and second graphene layers are held together by van der Waals force. The interlamination between the two graphene layers is relatively large. Therefore, it is easier for atoms to be intercalated between the graphene layers. Therefore, we mainly focus on the second case in this paper.
In this paper, employing ab initio density functional theoretical calculations, we show both the type of epitaxial graphene and the density of epitaxial graphene atoms on SiC(0001) can be deliberately controlled via decorating the buffer layer with specific atoms (i.e., F, Cl, O, N). More importantly, the fine tuning of the doping behavior from intrinsic n-type to charge neutrality to p-type and interface magnetism are achieved by increasing the adsorption concentration of F atoms on the buffer layer. For other atom doping cases, Cl doping has similar effects to the F doping, and N decoration is an effective way to induce a metal-semiconductor transition with the Dirac point moving but not disappearing, while O modification leads to a p-type doping. There have been experimental research studies of intercalation of F atoms [ 10 ] and the intercalation of O atoms, [ 40 ] but not on the intercalation of N atoms. As mentioned above, it is easier for atoms to be intercalated between the graphene layers. After intercalation, F atoms and O atoms may be adsorbed on the surface of the first graphene layer, while N atoms are more likely to link the first and second graphene layers. These are the atomic structures to be studied in this paper.
Our calculations are performed in the framework of density functional theory (DFT) as implemented in the PWSCF code. [ 41 ] The local spin density (LDA) approximation and projector augmented wave (PAW) potentials are used to describe electronic exchange–correlation and electron–core interactions, respectively. A kinetic cutoff of 400 eV is used in the plane wave expansion. The systems studied here consist of one, two or three carbon layers with Bernal stacking on 6H-SiC(0001) substrate. We adopt a 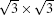
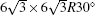
As shown in Fig.
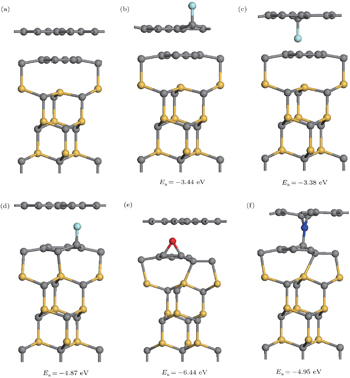
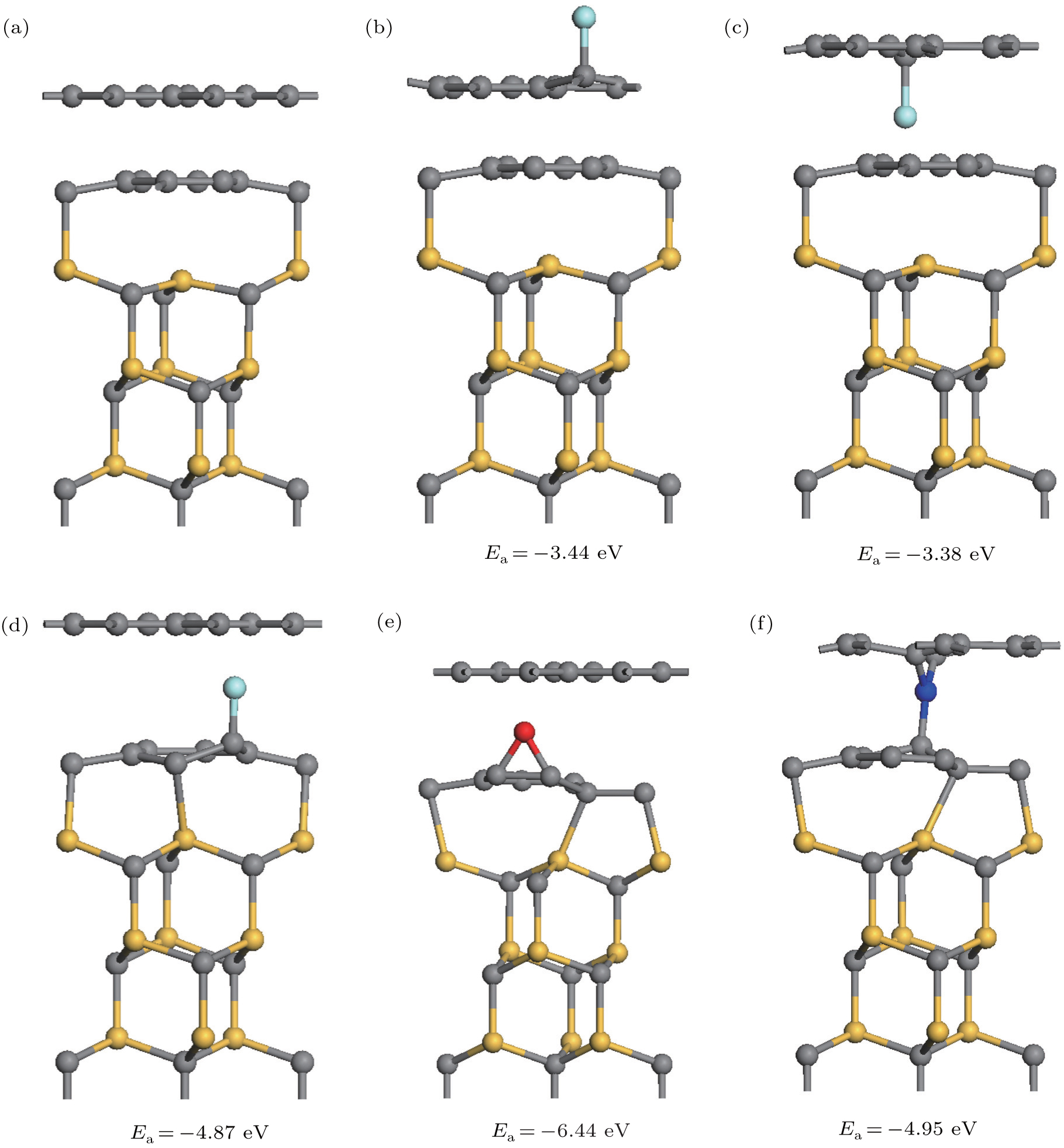 | Fig. 1. Optimized structures of perfect and doped epitaxial graphene on SiC(0001). (a) perfect system. (b)–(d) F decorated systems with different adsorption sites. (d) and (e) The stablest configurations of O, N decorated epitaxial graphene. |
Figure
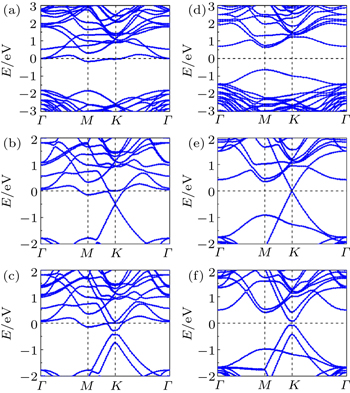
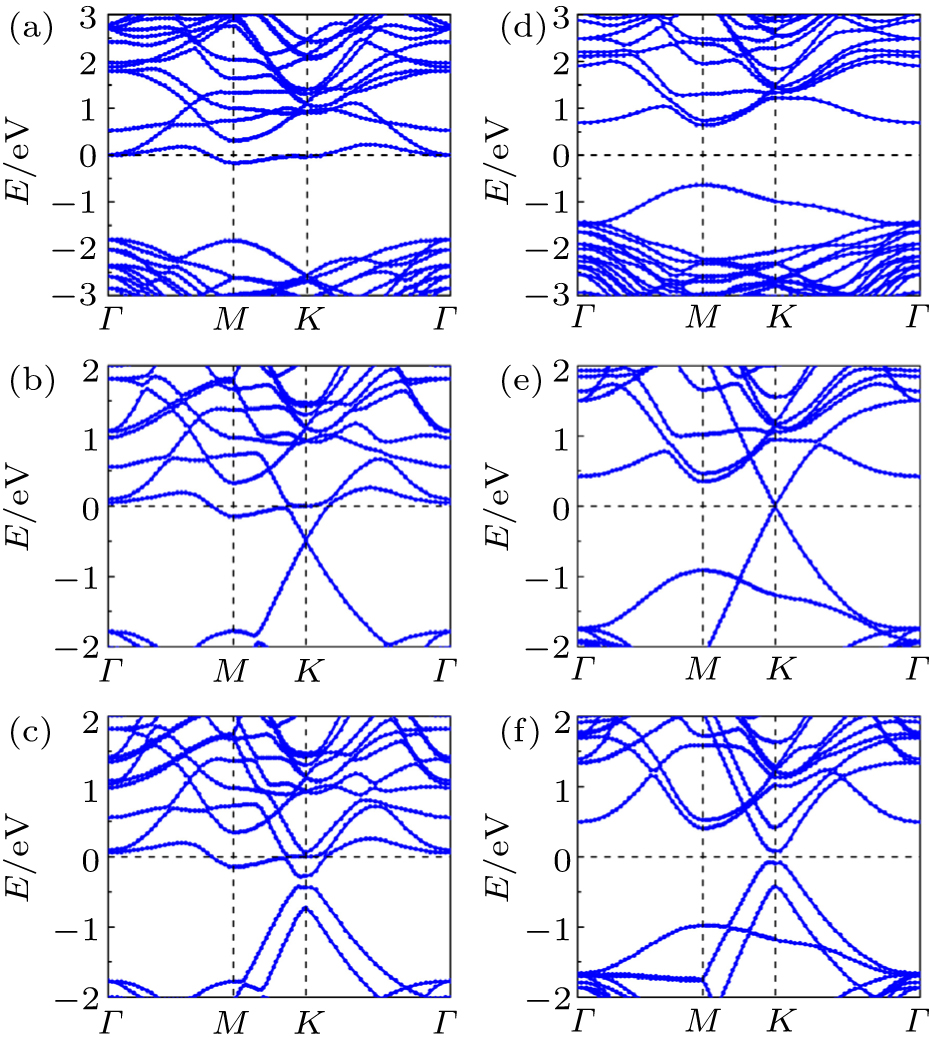 | Fig. 2. Band structures for one ((a) and (d)), two ((b) and (e)), three ((c) and (f)) carbon layers on the SiC(0001). Curves in panels (a)–(c) correspond to the perfect systems, while the curves in panels (d)–(f) correspond to the doped system with the first C layer decorated by one F atom. |
Next, we increase the adsorption concentration of F atoms until all the C l atoms in the buffer layer are adsorbed by F atoms, that is, six F adatoms in the unit cell. Unlike the case of single F decoration, one third of Si atoms at the interface are not saturated, like a perfect system. However, the DB related interface state of the six F atom doped system shows different behaviors from those of the perfect system. Figure
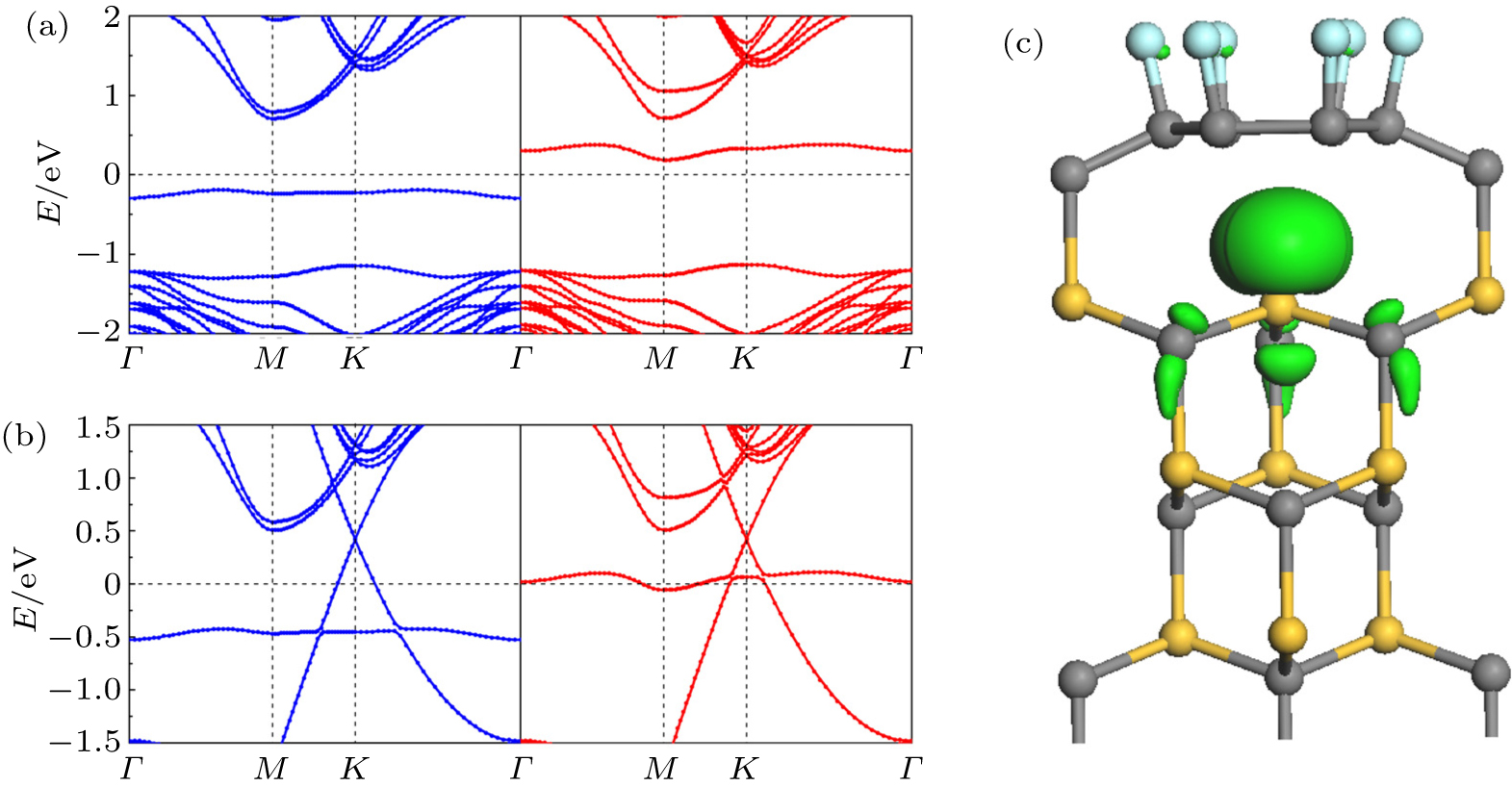 | Fig. 3. Band structures for (a) one and (b) two graphene layers on SiC(0001) with the first graphene layer doped by six F atoms. The blue and red curves represent the majority and minority spin, respectively. (c) Spin density ( ρ = ρ ↑ − ρ ↓) isosurfaces of the six F atoms decorated single layer epitaxial graphene. The isosurface is 0.025 e/Å 3 . |
So far, by analyzing the band structures, we know that the doping behaviors of the epitaxial graphene can be finely tuned from intrinsic n-type to charge neutrality to p-type by increasing the doping concentration of F atoms on the buffer layer. Then we turn to investigate how the F dopants tune the carrier properties and charge transfer. Taking the systems containing two graphene layers on the SiC for example, figures
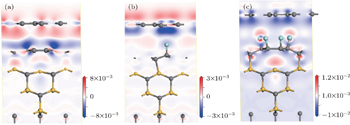
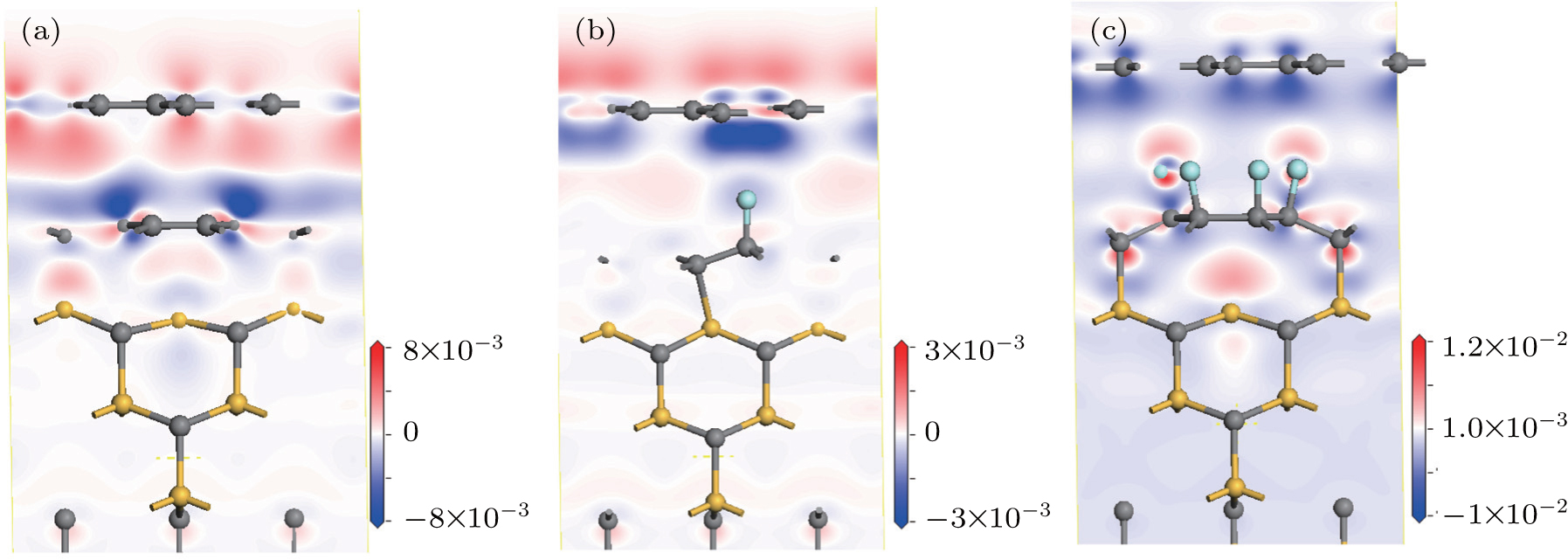 | Fig. 4. Contour plots of charge density difference of perfect system (a), single F atom decorated system (b), and six F atoms decorated system (c). Gray, yellow, and blue balls denote C, N, and F atoms, respectively. |
In addition, we also use Bader analysis, a measure of charge quantity localized around a given atomic site, to give a quantitative estimate of the charge variations of the F atoms and graphene layers. For the perfect system, the electron densities in the buffer layer and the top layer are ∼ 4.88 × 10 14 cm −2 and ∼ 3.85 × 10 13 cm −2 , respectively, consistent with its n-doped band structure. When the buffer layer is doped by an F atom, owing to the formation of another C–Si bond, an additional 0.44 electrons are transferred from the substrate. On the other hand, the F atom shows an onsite charge quantity of 7.64 e, which means that there are 0.64 holes injected into the graphene layer. This is due to the larger electronegativity of F than that of C. Thus, the electron density in the buffer layer is reduced to ∼ 4.50 × 10 14 cm −2 , showing a modest decrease. Most importantly, there is no charge transfer to the top graphene layer, which makes the Dirac point of the top layer lay exactly at the Fermi level. When the buffer is adsorbed by six F atoms, with each F atom accepting 0.59 e, enough holes are injected into the graphene layers, part of which can neutralize the electrons transferred from the substrate. Then the buffer layer and top layer transform into p-type, exhibiting hole densities of ∼ 8.59 × 10 14 cm −2 and ∼ 4.51 × 10 13 cm −2 , respectively.
Finally, we investigate the effects of the doping of other atoms (i.e., Cl, O, and N) on the buffer layer. Since Cl and F subordinate to the same main group, similar configurations and behaviors as discussed above are found. However, O doping on buffer layer shows quite different behaviors. As shown in Fig.
| Table 1. Manipulations of carrier type and gap in graphene by atoms (F, Cl, O, N) intercalations. F, or Cl, or O intercalation leads to a p-type doping in graphene, but cannot open the band gap of graphene. N intercalation cannot bring in external carriers but renders a band gap opening, and the band gap is 0.25 eV. . |
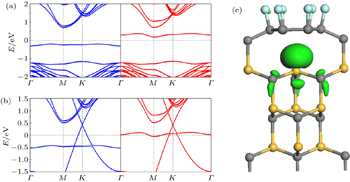
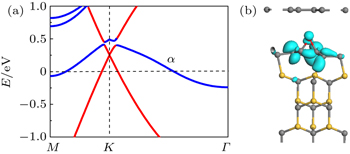
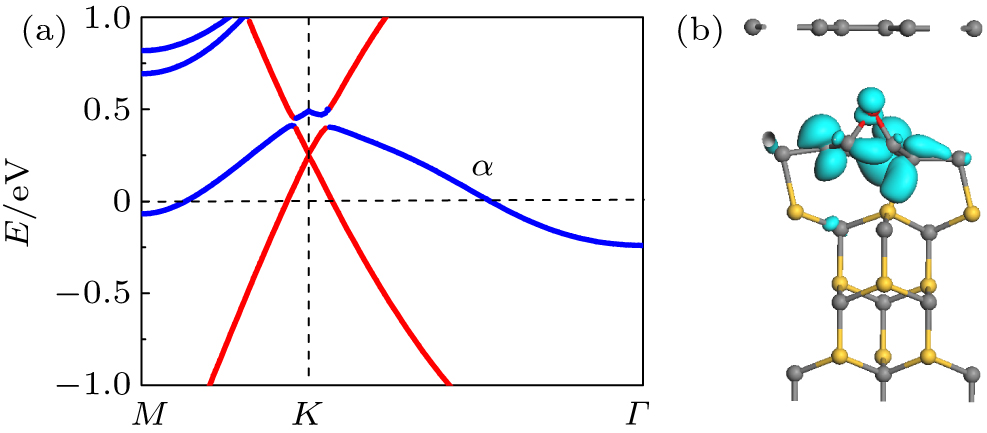 | Fig. 5. (a) Band structure of bilayer graphene with buffer layer doped by O on SiC(0001), with the top graphene energy bands being highlighted by red lines. (b) Partial charge density of the α band at the Γ point. The isosurface is 0.05 e/Å 3 . Gray, yellow, and red balls denote C, Si, and O atoms, respectively. |
Figure
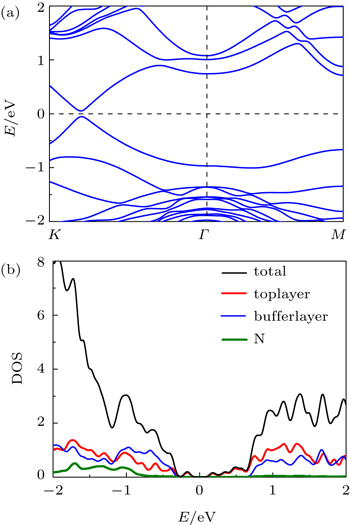
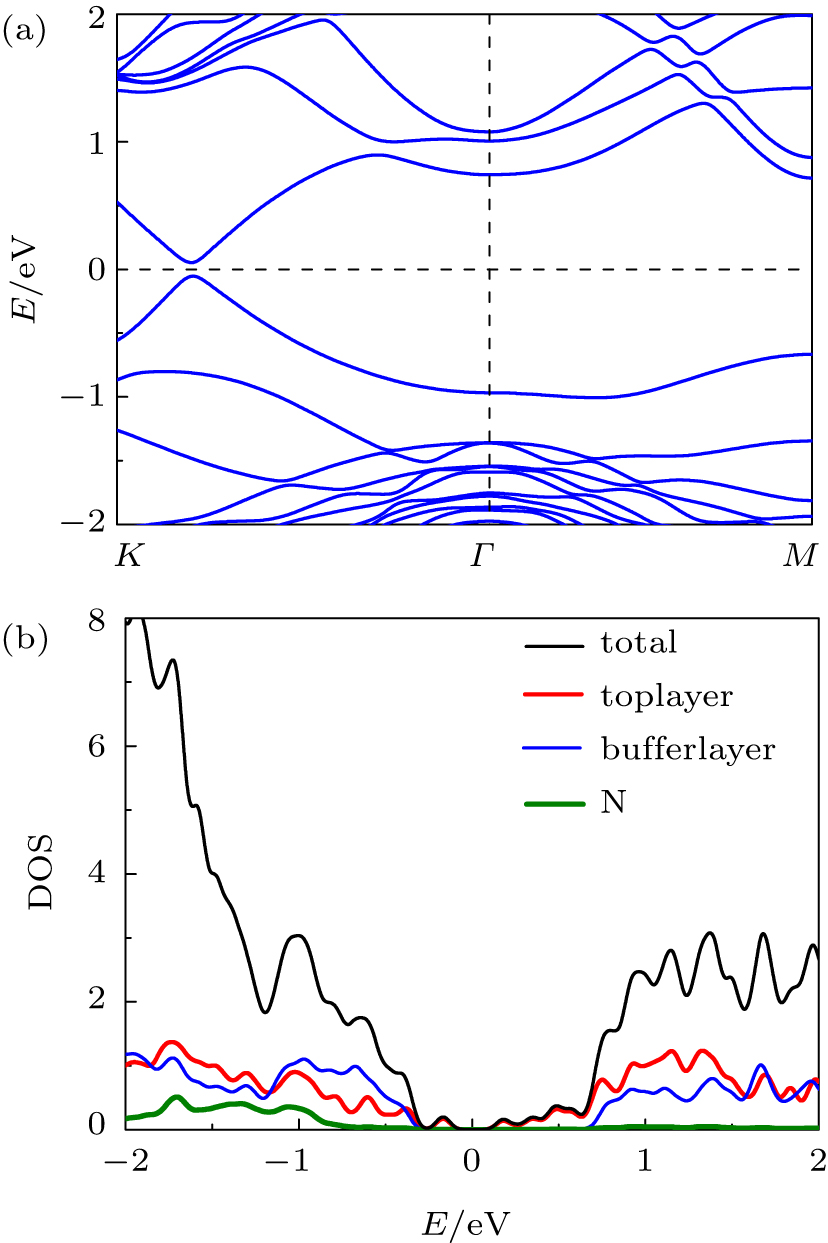 | Fig. 6. (a) Band structure of bilayer graphene on Si-terminated SiC substrate with N intercalation. (b) The curves of total density of states and local density of states versus energy for top C layer, buffer layer, and N. |
In this work, we investigate the band engineering and interface magnetism of epitaxial graphene on SiC(0001) by nonmetal atom doping. We find that its carrier type and density could be effectively manipulated by specific atoms (i.e., F, Cl, O, or N) decoration of the buffer layer. The fine tuning of the doping behavior from intrinsic n-type to charge neutrality to p-type and interface magnetism is achieved via increasing the adsorption concentration of F atoms on the buffer layer. Cl doping is similar to F doping. In addition, N decoration is also a good way to induce the metal-semiconductor transition with the Dirac point moving but not disappearing, while O modification can induce a p-type doping behavior. Our findings establish the potential for epitaxial graphene applications in nanoscale electronic and spintronic devices. Meanwhile, the mentioned ideas of intercalation in the graphene/substrate system can also be applied to similar systems such as silicone/substrate.
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 | |
| 23 | |
| 24 | |
| 25 | |
| 26 | |
| 27 | |
| 28 | |
| 29 | |
| 30 | |
| 31 | |
| 32 | |
| 33 | |
| 34 | |
| 35 | |
| 36 | |
| 37 | |
| 38 | |
| 39 | |
| 40 | |
| 41 | |
| 42 |