





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Label-free surface-enhanced infrared spectro-electro-chemical analysis of the Redox potential shift of cytochrome c complexed with a cardiolipin-containing lipid membrane of varied composition
[Liu Li, Wu Lie, Zeng Li, Jiang Xiu-E† ]
]
]
|
|


†Corresponding author. E-mail: jiangxiue@ciac.ac.cn
*Project supported by the National Natural Science Foundation of China (Grant Nos. 91227114, 21322510, and 21105097), the China Postdoctoral Science Foundation (Grant No. 2013M530998), the Natural Science Foundation of Jilin Province, China (Grant No. 201215092), and the President Funds of the Chinese Academy of Sciences.
In this study, a lipid membrane was fabricated by fusing cardiolipin-phosphatidylcholine (CL_PC, 1:4) vesicles onto a hydrophobic surface of 1-dodecanethiol (DT) preadsorbed on a nanostructured gold film. By changing the concentration of the DT adsorption solution, we constructed a series of CL_PC-DT bilayers with different hydrophobicity to study the effects of lipid membrane characteristics on the adsorption conformation of cytochrome c (Cyt c). Electrochemical analysis showed that the formal potential is 0.24 V for Cyt c-CL_PC-DT(10), 0.2 V for Cyt c-CL_PC-DT(20), and 0.16 V for Cyt c-CL_PC-DT(40) — a gradual positive shift with the decreasing DT concentration — relative to the potential of native cyt c (0.02 V). Potential-induced surface-enhanced infrared adsorption difference spectroscopy revealed that the gradual positive shift of the formal potential of CL-bound cyt c is determined by the environment with the gradually lowered dielectric constant for the heme cofactor in CL-bound cyt c (Fe3+).
Apoptosis is a carefully regulated biochemical death program through which irreparably damaged, genetically altered, or unwanted cells are eliminated. To date, general principles of apoptosis have been deciphered, [1] but the specific details of triggering events remain less clear. It is generally accepted that the intrinsic pathway of apoptosis is triggered by a release of cytochrome c (cyt c) from mitochondria into cytoplasm, where cyt c induces a cascade of biochemical events.[2– 5] Interactions of cyt c with cardiolipin (CL), a mitochondria-specific phospholipid, are important for this release.[6] The binding of CL activates a peroxidase activity in cyt c by promoting protein unfolding.[7] The peroxidase activity targets CL itself, and CL peroxidation in turn contributes to permeabilization of the mitochondrial membrane.[8]
To identify the mechanism of the apoptosis induced by the cyt c-CL complex, some advanced techniques and strategies have been used to analyze the complex-induced conformation change of the protein and the redox properties of the complex.[5, 9, 10] Perhirin et al. and Basova et al. independently uncovered a negative shift of the formal potential of cyt c after it binds to CL-containing liposomes; these researchers reported hydrophobic characteristics of this binding and the peroxidase activity of this complex.[9, 10] These electrochemical assays cannot further elucidate the causes of these events in terms of the solvent involvement and the conformational changes of the protein. To find a possible correlation of the structural features with the apoptogenic effects of the cyt c-CL complex, Patriarca et al. chose to analyze the complex of H26Y with CL and identified the structural properties of partial unfolding of the protein that are required for peroxidase activity.[5] This strategy cannot eliminate the false signals from the structural perturbations after a mutation and labeling. Thus, a label-free and sensitive method is urgently needed for such experiments.
Surface-enhanced infrared adsorption spectroscopy (SEIRAS) is a label-free, in situ, and real-time detection method for studies on the molecular structure and secondary structure of proteins in a superthin layer on a nanogold or nanoplatinum surface.[11– 15] In combination with electrochemistry, potential-induced SEIRA difference spectroscopy appears to be a suitable method for analysis of interactions between redox proteins, between a redox protein and lipid membrane, or between a protein and solvent for elucidation of the biological functions or mechanisms of action of various proteins at the molecular level.[16– 19] First, SEIRAS can detect in situ in real time the formation of lipid membranes and subsequent adsorption of a protein; these data can serve as a reliable experimental basis for further studies. Second, potential-induced SEIRA difference spectroscopy can precisely detect small changes in the secondary structure and in component groups of a redox protein by subtracting the background spectrum (in the reduced state) from the sample spectrum (in the oxidized state).[16– 19] Using the surface selection rule and optical near-field effect, [20] researchers can derive the conformational change and orientation of the protein after interaction with other proteins or lipid membranes.[16, 18] Third, potential-induced SEIRA difference spectroscopy is still a good method for analysis of the protein-solvent interaction via detection of the change in the degree of hydration of the adsorbing protein and the adsorbed lipid membrane during the redox process. It is well known that dynamic proteinsolvent or nucleic acid-solvent interactions are essential for life processes.[21] For example, the extent to which a protein matrix shields a redox site from solvent water molecules determines the activation barrier for electron transfer and thus the formal potential of the protein.[22] Furthermore, the interaction between a solvent and DNA is proportional to their contact area and determines the quasispherical or stretched configuration in the solvent and eventually translocation of the polymer chain.[23] Nevertheless, the empirical research on these characteristics is limited by the current methods and skills although some research has been carried out by means of computational simulations.[21] Thus, SEIRAS analysis of protein-solvent interactions is a worthwhile approach.
In this work, we constructed solid-supported lipid membranes of CL_ PC via vesicle fusion onto gold electrodes precoated with DT to mimic the outer leaflet of the inner membrane of mitochondria. By changing the concentration of the DT adsorption solution, we fabricated a series of bilayer membranes with different hydrophobicity and solvent transport performance to clarify the effect of lipid membrane characteristics on the adsorption properties of cyt c. SEIRAS was used to monitor formation of the series of lipid bilayers and subsequent adsorption of cyt c. The electrochemical analysis and potential-induced SEIRA difference spectroscopy of Cyt c-CL_ PC-DT, as compared to those of Cytc-MUA (11- mercaptoundecanoic acid), were also conducted. We found that the hydrophobic interaction between cyt c and the lipid membranes indeed occurred. With the decreasing DT concentration, the gradually decreasing dielectric constant of the environment for the heme cofactor in CL-bound cyt c (Fe3+ ) and different adsorption orientation determine the gradual positive shift in the formal potential of CL-bound cyt c. Cyt c did not show partial unfolding after binding to CL and therefore peroxidase activity was not induced, in contrast to other studies.[5, 7] Nevertheless, our results may provide further insights into the mechanism of early apoptosis induced by the cyt c-CL complex.
The flat surface of a triangular silicon prism was polished with 1.0-μ m Al2O3 slurry and then washed thoroughly with pure water. The clean silicon substrate was pretreated by immersion in a 40-wt% aqueous solution of NH4F for 1 min. A thin gold film was applied to the pretreated triangular silicon prism by chemical deposition.[24] In brief, the flat surface of the pretreated silicon substrate was exposed to a 1:1:1 (v/v/v) mixture of (i) 0.03-M NaAuCl4, (ii) 0.3-M Na2SO3 + 0.1 M Na2S2O3 + 0.1 M NH4Cl, and (iii) 2.5-vol% HF solution for 90 s. After rinsing with water, the silicon prism was transferred into a 0.1-M H2SO4 solution. The gold film surface was cleaned by several oxidation-reduction cycles in the range 0.1 V– 1.4 V (versus Ag/AgCl). After electrochemical cleaning, the nanostructured gold-coated silicon prism was mounted into a poly(trifluorochloroethylene) cell, and the reflected signals were detected at the gold-silicon thin film-solution interface.
This procedure was described elsewhere.[25] Phosphatidylcholine (PC, Sigma) was dissolved in chloroform, and cardiolipin (CL) from bovine heart (ethanol solution; Sigma- Aldrich) was added to the PC-containing chloroform solution in the molar ratio 1:4 (CL:PC). The solvent was evaporated under a nitrogen stream to initiate formation of a thin lipid layer, then placed in a vacuum chamber for 2 h. Ten microliters of phosphate buffer (PB, pH 7.0) were added to obtain the final concentration of CL_ PC of 1 mg/mL and then sonication at 40 ° C for 1 h yielded a clear solution. The CL_ PC vesicles were thus obtained, and the vesicle solution was always used within 24 h after preparation.
First, a reference spectrum was recorded (blank, ethanol) on the nanostructured gold film. Simultaneously with the addition of a 10, 20, or 40-μ M ethanol solution of 1-dodecanethiol (DT, Sigma-Aldrich) onto the gold film, a sample spectrum was acquired at the spectral resolution of 4 cm− 1. Then, a series of DT-gold films were washed with ethanol, water, and 10-mM PB in that order. After that, 500 μ L of 10-mM PB was added to the DT-gold films and scanned to obtain reference spectra again. The sample spectra were recorded after 1-mg/mL CL_ PC vesicle solution was added and kept for 3 h to enable formation of a planar membrane on the DT-gold surface via vesicle fusion. Then, after exposure of a series of CL_ PC-DT-gold surfaces to 2 μ M cyt c in 10-mM PB, the adsorption of cyt c was monitored (with 10-mM PB subtracted as a background) during formation of Cyt c-CL_ PC-DT(10), Cyt c-CL_ PC-DT(20), or Cyt c-CL_ PC-DT(40). Preparation of a Cyt c-MUA-gold film has been described elsewhere.[16] For each spectrum, 512 scans were performed at the spectral resolution of 4 cm− 1.
The gold-silicon prism was tightened in the electrochemical cell (the poly[trifluorochloroethylene] cell) with a Viton O-ring. A copper plate serves here as the electric contact for the gold film. The gold-silicon substrate was used as a working electrode. A platinum wire and a Ag/AgCl electrode with a saturated KCl solution served as counter and reference electrodes, respectively. Cyclic voltammograms were recorded on a CHI 830A electrochemical workstation (CH Instruments, Austin, TX); 10-mM PB was purged with highly pure nitrogen gas for at least 10 min prior to electrochemical measurements to remove the dissolved oxygen. For the series of the Cyt c-CL_ PC-DT films and the Cyt c-MUA (11- mercaptoundecanoic acid) film, a reference SEIRA spectrum was recorded at a reduction potential, where the adsorbed cyt c was fully reduced, and a sample spectrum was acquired at an oxidation potential, when the adsorbed cyt c was fully oxidized. The whole procedure was repeated nine times, and these difference spectra were averaged to improve the signalto- noise ratio.
The adsorption of 10, 20, or 40-μ M DT in ethanol onto a nanostructured gold surface was monitored by SEIRA spectroscopy, and the individual saturation adsorption spectra are shown in Fig. 1. The roughness factor of these nanostructured gold films (according to the Au-oxide reduction charge density method) was basically the same (data not shown). Therefore, the values of their surface enhancement factor can be considered roughly equal. Thus, the difference among the adsorption spectra should reflect the change (as a function of DT concentration) in the coverage of the DT monolayer and in the orientation of its alkyl chains. As shown in Fig. 1, the saturated coverage of the DT monolayer increases with the DT concentration. The asymmetric, ν as(CH2), and symmetric, ν s(CH2), stretching vibrations occurred at 2925 cm− 1 and 2853 cm− 1 for the DT monolayer derived from 10-μ M DT in ethanol (DT(10)), at 2922 cm− 1 and 2852 cm− 1 for the DT(20) monolayer, and at 2921 cm− 1 and 2851 cm− 1 for the DT(40) monolayer. The position of the CH2 asymmetry vibration can be used as an indicator of the packing of alkyl chains in a film.[26, 27] When the band appears at 2916 cm− 1– 2918 cm− 1, it is likely that the alkyl chains assume an alltrans- cis zigzag conformation. A shift of this band to a greater wavenumber indicates the existence of the gauche conformation or disarray of the alkyl chains.[26, 27] Here, the greater the DT concentration, the more densely and orderly packed the alkyl chains are in the DT monolayer. On the other hand, the ratio of intensity of ν as(CH2) to that of ν s(CH2) is 2.33, 2.64, and 2.92 for the DT(10), DT(20), and DT(40) monolayers. In a transmission spectrum, a much higher intensity ratio of ν as(CH2) to ν s(CH2) indicates that the alkyl chains are nearly perpendicular to the film plane.[26, 27] In SEIRAS, such an intensity ratio can be further amplified because, here, only those vibrations are IR-active and greatly enhanced that have a dipole moment that changes perpendicularly to the (local) surface.[20] Thus, the greater the DT concentration, the more perpendicular (approximately) to the film surface the alkyl chains are in the DT monolayer.
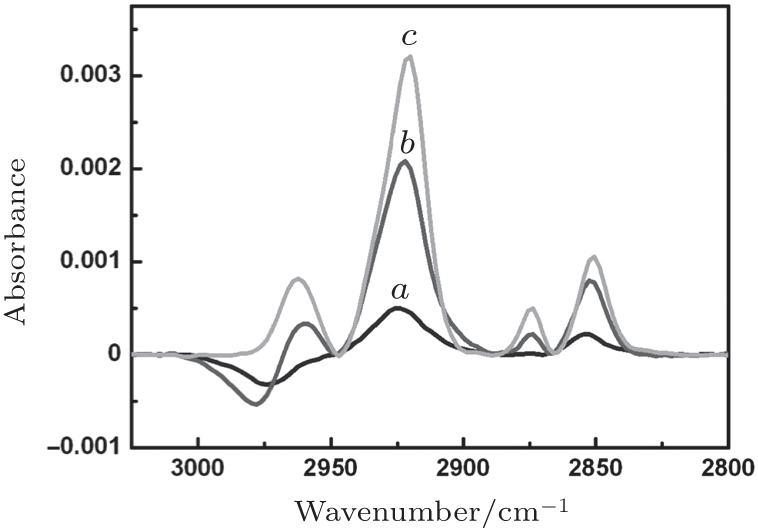 | Fig. 1. Surface-enhanced infrared adsorption spectroscopy (SEIRAS) monitoring of the adsorption of 10-μ M (curve a), 20-μ M (curve b), or 40-μ M (curve c) 1-dodecanethiol (DT) in ethanol onto a nanostructured gold surface. All spectra are at their individual time point of saturation adsorption. |
The formation of lipid membranes by spreading and fusion of CL_ PC (1:4) vesicles on the series of DT monolayers was also monitored by SEIRAS. The individual spectra of saturation adsorption and their second derivatives are shown in Fig. 2. These adsorption spectra were obtained using the spectra of the DT monolayers as a reference. Here, positive and negative bands represent contributions of the species bound to and removed from the DT-gold surface, respectively. The positive bands between 3050 cm− 1 and 2800 cm− 1 and between 1780 cm− 1 and 1690 cm− 1 are assigned to C– H stretching modes of the alkyl chains of CL_ PC and C= O stretching bands of the ester groups of CL_ PC, respectively. The broad negative bands of 1690 cm− 1– 1570 cm− 1 were caused by the removal of the water molecules (close to the DT monolayers) that were replaced by CL_ PC molecules. Judging by the intensity of both C– H and C= O stretching bands, the coverage of the CL_ PC layer increases with the concentration of the DT adsorption solution, namely with the coverage of its underlying DT monolayer. Thus, as the DT concentration increases, the CL_ PC-DT bilayer is formed with the alkyl chains packed more and more densely. According to the intensity of the negative band, more and more water molecules are removed from the DT monolayer during the formation of the CL_ PC-DT bilayer as the concentration of the DT adsorption solution increases, and thus the hydrophobicity of the bilayer increases. On the other hand, the position of the C= O stretching vibration can be identified by means of the second derivatives of the spectra (Fig. 2, inset). The C= O vibration band of CL_ PC-DT(10) is predominant at 1725 cm− 1; in both CL_ PC-DT(20) and CL_ PC-DT(40), those bands are observed at 1744 cm− 1 for the majority and at 1725 cm− 1 for the minority. The bands of 1725 cm− 1 and at 1744 cm− 1 can be assigned to the C= O stretching modes of CL_ PC in lateral hydrogen-bonding (H-bonding) mode and without it, respectively. In the film containing C= O mainly in H-bonding mode, the alkyl chains are aligned at the least dense packing and with the smallest number of water molecules removed during the formation of the bilayer. For coexistence of the two modes, the following dependence is true: the greater the coverage of CL_ PC and DT, the more water molecules are removed during the formation of the bilayer.
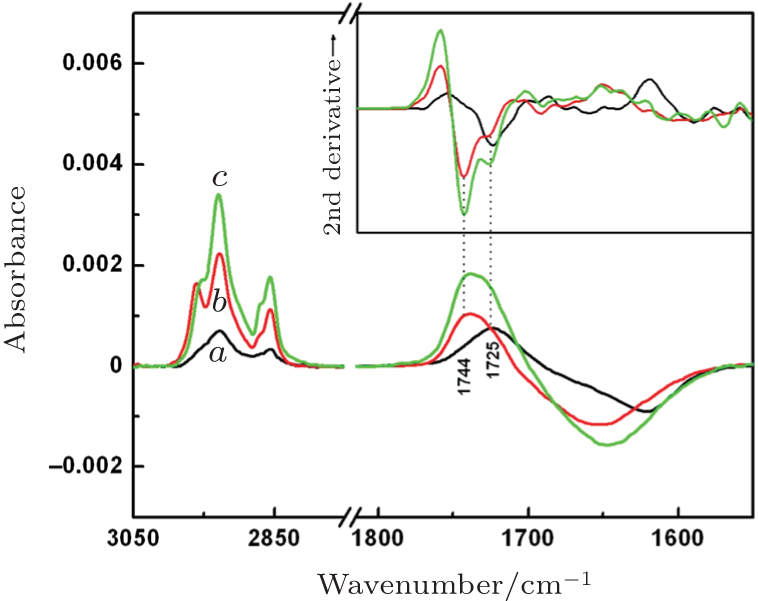 | Fig. 2. Surface-enhanced infrared adsorption spectroscopy (SEIRAS) monitoring of the formation of cardiolipin-phosphatidylcholine (CL_ PC) lipid membranes on a series of 1-dodecanethiol (DT)-gold surfaces via vesicle fusion of 1 mg/mL CL_ PC (1:4) vesicles onto DT(10) (curve a), DT(20) (curve b), and DT(40) (curve c) monolayers. The inset shows the second-derivative spectra. All spectra are at their individual time point of saturation adsorption. |
Figure 3 shows SEIRA spectra of cyt c adsorbed onto each of our CL_ PC-DT bilayers at their individual adsorption time point. In each spectrum, two major peaks, at 1659 cm− 1 and 1551 cm− 1, are seen, which are assigned to amide I mode (mainly C= O stretching vibration of the peptide backbone) and to amide II mode (coupled mode of the NH in-plane bending, CN stretching, and Cα C stretching vibration), respectively. All three CL-bound cyt c have a similar band contour of amide I to native cyt c in the case of Cyt c-MUA-gold.[15] Gaussian curve-fitting analysis of the amide I band shows that the secondary structure of CL-bound cyt c undergoes only a small change relative to that of native cyt c (Table 1). The proportion of β -sheets and random amino acid sequences shows a slight increase at the expense of α -helices and β -turns.
In this case, the surface coverage is derived from the total area of the amide I and II bands. Figure 3 also shows that the coverage ratio of cyt c is 1:2.1:2.5 for Cyt c-CL_ PC-DT(10), Cyt c-CL_ PC-DT(20), and Cyt c-CL_ PC-DT(40), respectively. According to Fig. 2, the coverage of cyt c is proportional to the coverage of its underlying CL_ PC-DT bilayer and to the displacement of water during formation of this bilayer. Thus, it can be deduced that the hydrophobic interaction occurs between cyt c and the CL_ PC-DT bilayer. After the initial electrostatic recognition, the hydrophobic interaction between cyt c and CL_ PC-DT caused minor changes in the conformation of cyt c. On the other hand, the amide I/II intensity ratio is 1.77 for Cyt c-CL_ PC-DT(10), 1.42 for Cyt c-CL_ PC-DT(20), and 1.34 for Cyt c-CL_ PC-DT(40). The intensity ratio of the amide I/II bands has also been used as a tool useful for qualitative analysis of orientation changes.[16] It is known that for a protein helix, if dipole moment of its amide I band is perpendicular to the local surface, then dipole moment of its amide II mode is parallel to the surface.[16] According to the surface selection rule of SEIRAS, [20] a greater ratio of amide I/II intensity indicates that a greater proportion of α -helices adopt an orientation perpendicular to the film surface. Thus, in this case, a slightly greater proportion of α -helices is positioned perpendicularly to the film surface in Cyt c-CL_ PC-DT(20) than in Cyt c-CL_ PC-DT(40), and an obviously greater proportion in the Cyt c-CL_ PC-DT(10) film than in the other two films.
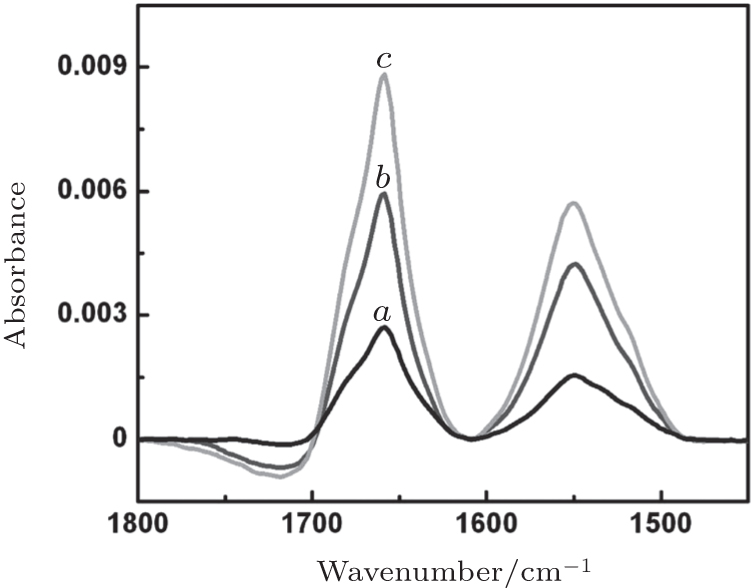 | Fig. 3. Surface-enhanced infrared adsorption (SEIRA) spectra of cytochrome c adsorbed onto the films CL_ PC-DT(10) (curve a), CL_ PC-DT(20) (curve b), or CL_ PC-DT(40) (curve c) at their individual time point of saturation adsorption. CL_ PC: cardiolipin-phosphatidylcholine bilayer, DT: 1-dodecanethiol. |
| Table 1. Secondary-structure content of cytochrome c as estimated by surface-enhanced infrared adsorption spectroscopy (SEIRAS). |
These electrochemical characteristics were analyzed in comparison with that of the Cyt c-MUA film in 10-mM PB (pH 7) at the scan rate of 50 mV/s, as shown in Fig. 4. A pair of well-defined redox peaks for Cyt c-CL_ PC-DT(10) was observed at 0.13 V and 0.35 V with the formal potential (Ef) of 0.24 V and the peak separation (Δ E) of 220 mV, also a pair at 0.10 V and 0.30 V with Ef = 0.2 V and Δ E = 200 mV for Cyt c-CL_ PC-DT(20), and a pair at 0.11 V and 0.21 V with Ef = 0.16 V and Δ E = 100 mV for Cyt c-CL_ PC-DT(40). Relative to native cyt c (Ef = 0.02 V and Δ E = 30 mV), CL-bound cyt c showed a gradually positively shifted Ef and greater Δ E with the decreasing concentration of the DT adsorption solution. It is likely that for each CL-bound cyt c, it is easier to undergo reduction and more difficult to undergo oxidation in comparison with native cyt c. With the decreasing DT concentration, the oxidation potential of CL-bound cyt c shifts positively and its reduction potential undergoes only a small change, suggesting that oxidation became more difficult, while the ease of the reduction reaction did not change significantly.
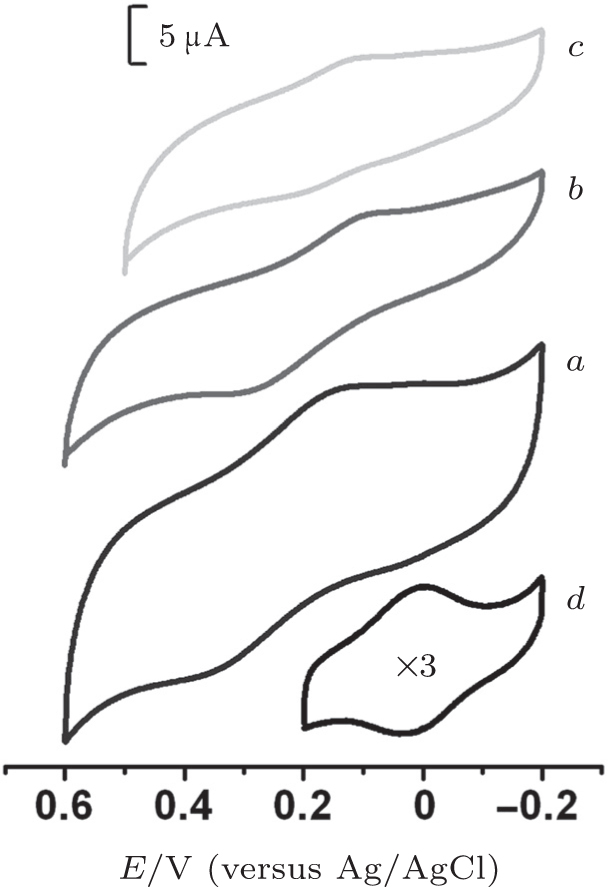 | Fig. 4. Cyclic voltammograms of the complexes Cyt c-CL_ PC-DT(10) (curve a), Cyt c-CL_ PC-DT(20) (curve b), Cyt c-CL_ PC-DT(40) (curve c), and Cyt c-MUA (curve d) in 10-mM phosphate buffer (pH 7) at the scan rate of 50 mV/s. CL_ PC: cardiolipin-phosphatidylcholine bilayer, DT: 1-dodecanethiol, MUA: 11-mercaptoundecanoic acid. |
Although the reversibility gradually worsened, the faradic current of Cyt c-CL_ PC-DT increased with the decreasing DT concentration. Because the perpendicular orientation of α -helices in the film containing adsorbed cyt c is crucial for the electrochemical activity of cyt c, [16] the response current in this study is proportional to not only the coverage of cyt c but also the orientation of cyt c (the amide I/II intensity ratio).
Figure 5 shows these spectra of CL-bound cyt c on the series of the Cyt c-CL_ PC-DT films and spectra of native cyt c on the Cyt c-MUA film in 10-mM PB. The reference spectra were recorded at − 0.1 V in all cases, where the adsorbed cyt c was fully reduced. The sample spectra were acquired at 0.1 V for Cyt c-MUA, at 0.35 V for Cyt c-CL_ PC-DT(40), at 0.4 V for Cyt c-CL_ PC-DT(20), and at 0.5 V for Cyt c-CL_ PC-DT(10), where all the adsorbed cyt c molecules are fully oxidized.
Figure 5 also shows the potential-induced difference spectra of the underlying CL_ PC-DT bilayers (grey), in which the broad water bands at ∼ 3500 cm− 1 increased in intensity with the decreasing DT concentration. This finding is suggestive of rearrangement of the interfacial water molecules that are H-bonded to the polar headgroups of CL_ PC.[18] The adsorption of cyt c further promoted the potential-dependent increase in the total hydration of the Cyt c-CL_ PC-DT film through a change in hydration of the protein itself during the redox process. This phenomenon is indicated by the difference in the intensity of negative peaks at ∼ 3500 cm− 1 between the film with adsorbed cyt c and the underlying bilayer or monolayer (Δ I). The intensity ratio of Δ I to 1693-cm− 1 vibration further reveals the contribution of each adsorbed cyt c molecule to such an increase in hydration. (The 1693-cm− 1 vibration that is assigned to the type III β -turns of the protein denotes the coverage of cyt c.[16, 18]) It can be deduced that either CL-bound cyt c (Fe2+ ) is more hydrated or CL-bound cyt c (Fe3+ ) has greater hydrophobicity. It is known that native cyt c (Fe2+ ) has a degree of hydration similar to that of native cyt c (Fe3+ ).[28] In addition, CL-bound cyt c (Fe2+ ) should have a similar conformation and thus a hydration degree similar to that of native cyt c (Fe2+ ). This is because the reduction of partially unfolded cyt c (Fe3+ ) always causes structural folding that involves formation of the native conformation from an MG (Molten Globule) intermediate species, owing to the greater affinity of Met80-Fe2+ .[29– 31]
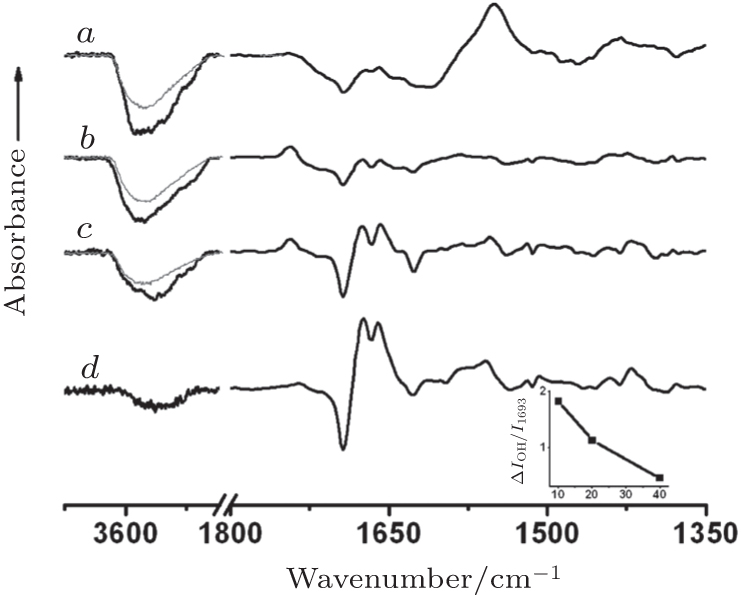 | Fig. 5. The potential-induced surface-enhanced infrared adsorption (SEIRA) difference spectra of cyt c-containing (black curve) and blank films (grey curve) CL_ PC-DT(10) (curve a), CL_ PC-DT(20) (curve b), CL_ PC-DT(40) (curve c), and Cyt c-MUA (curve d) in 10-mM PB (pH 7), with the reference potential − 0.1 V and the sample potential 0.35 V (curve a), 0.4 V (curve b), 0.5 V (curve c), and 0.1 V (curve d). The arrow corresponds to absorbance Δ A of 5 × 10− 4. Inset: the ratio Δ IO− H/I1693 versus 1-dodecanethiol (DT) concentration, where Δ IO− H is the difference in the intensity of negative peaks at ∼ 3500 cm− 1 between the complexes Cyt c-CL_ PC-DT and CL_ PC-DT, and I1693 is the intensity at 1693 cm− 1. CL_ PC: cardiolipin-phosphatidylcholine bilayer, MUA: 11-mercaptoundecanoic acid. |
Thus, in this case, CL-bound cyt c (Fe3+ ) may have greater hydrophobicity and drive more water molecules off than native cyt c (Fe3+ ) can. Namely, the heme cofactor in CL-bound cyt c (Fe3+ ) is exposed to an environment with a lower dielectric constant in comparison with the heme in native cyt c (Fe3+ ), whereas the heme cofactor is in almost the same environment (in terms of the dielectric constant) in CLbound cyt c (Fe2+ ) and native cyt c (Fe2+ ). These conditions may cause the positive shift of Ef of CL-bound cyt c.[32, 33] In other words, it is the change in solvent accessibility to the heme that causes the positive shift of Ef in CL-bound cyt c; the solvent-induced change in the reaction pathway does not contribute to this shift.[34] The inset of Fig. 5 shows that the ratio of Δ I to 1693-cm− 1 vibration increases with the decrease in DT concentration, indicating that CL-bound cyt c (Fe3+ ) may gradually become more hydrophobic and drive more and more water molecules off with the decreasing DT concentration. The environment with the gradually lowered dielectric constant for the heme cofactor in CL-bound cyt c (Fe3+ ) and a similar environment for CL-bound cyt c (Fe2+ ) could be the root cause of the positive shift of Ef in CL-bound cyt c with the decreasing DT concentration. This notion is supported by the evidence that the reduction potential of CL-bound cyt c shows almost no change, while its oxidization potential shifts positively with the decreasing DT concentration.
Overall, the environment with the lowered dielectric constant for the heme cofactor may be caused by the slight increase in the proportion of β -sheets and random amino acid sequences at the expense of α -helices and β -turns in CL-bound cyt c (Table 1). The hydrophobic interaction with the CL_ PCDT bilayer induced the conformational change of cyt c. Nevertheless, because of the gradually positively shifted Ef of CLbound cyt c, the following dependence is true: the weaker the hydrophobic interaction, the more favorable this interaction is for formation of the environment with the low dielectric constant. Probably, after electrostatic and hydrophobic adsorption onto CL_ PC-DT, molecules of cyt c may still have to escape from the partial hydrophobic interaction to come close to each other so that the dimer-like packing could cause the eventual formation of the environment with the low dielectric constant, [33] as shown in Fig. 6. The two heme cofactors in the dimer-like packing can equally accept an electron from and donate an electron to the electrode surface, in contrast to cytochrome c oxidase where an electron is transferred from heme a to the binuclear center.[35]
 | Fig. 6. Surface adsorption models of cyt c according to our results on surface-enhanced infrared adsorption (SEIRA) differences and previously reported results.[16, 18] Left panel: Cyt c adsorbed on the MUA-gold surface with high polarizability. Right panel: Cyt c adsorbed on the CL_ PC-DT-gold surface with low polarizability. CL_ PC: cardiolipin-phosphatidylcholine bilayer, DT: 1-dodecanethiol, MUA: 11-mercaptoundecanoic acid. |
From the energy point of view, the redox-dependent water rearrangement is the major determinant of the total reorganization energy (λ ) for cyt c: the energy required to distort the equilibrium nuclear configuration of the reactants toward the equilibrium configuration of products before electron transfer.[36] In this case, the greatly enhanced change in the redox-induced water rearrangement for CL-bound cyt c inevitably increases λ of its redox process and eventually causes the increase in Δ Ep and the irreversibility of CL-bound cyt c in comparison with native cyt c.[28, 36, 37] With the decreasing DT concentration, the increasing change in the redox-induced water rearrangement contributes to the gradual increase in Δ Ep and the irreversibility of CL-bound cyt c. In Fig. 5, one can see that intensity of bands for the oxidized state of CL-bound cyt c is much weaker than that of the reduced state across the entire spectral range. This result indicates that CL-bound cyt c (Fe3+ ) may be farther away from the surface compared to CL-bound cyt c (Fe2+ ).[18] Such redox-dependent floating of a protein may actually facilitate its dehydration and rehydration processes and is suggestive of the weak interaction between the protein and the lipid membrane. Thus, we can conclude that irreversibility of the protein-electrode processes is determined by the proximity of CL-bound cyt c to and disengagement from the electrode surface prior to or after electron transfer, accompanied by the reorientation of its heme plane and the reorganization of its protein matrix under the influence of the electric field.
After binding to a CL_ PC-DT film, cyt c acquires a gradually positively shifted formal potential in 10-mM PB (pH 7.0) with the decreasing DT concentration (in the film; according to experiments with different films). Potential-induced SEIRA difference spectroscopy shows that an environment with the gradually lowered dielectric constant for the heme cofactor in CL-bound cyt c (Fe3+ ) may be the root cause of the positive shift of the formal potential for CL-bound cyt c with the decreasing DT concentration. The increasing reorganization free energy shows a gradual increase in peak separation and the irreversibility of CL-bound cyt c with the decreasing DT concentration. Proximity of CL-bound cyt c to and disengagement from the electrode surface prior to or after electron transfer— accompanied by reorientation of its heme plane and by reorganization of its protein matrix in the electric field — confirm the irreversibility of the protein– electrode processes.
| 1 |
|
| 2 |
|
| 3 |
|
| 4 |
|
| 5 |
|
| 6 |
|
| 7 |
|
| 8 |
|
| 9 |
|
| 10 |
|
| 11 |
|
| 12 |
|
| 13 |
|
| 14 |
|
| 15 |
|
| 16 |
|
| 17 |
|
| 18 |
|
| 19 |
|
| 20 |
|
| 21 |
|
| 22 |
|
| 23 |
|
| 24 |
|
| 25 |
|
| 26 |
|
| 27 |
|
| 28 |
|
| 29 |
|
| 30 |
|
| 31 |
|
| 32 |
|
| 33 |
|
| 34 |
|
| 35 |
|
| 36 |
|
| 37 |
|