




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Triphenylene adsorption on Cu(111) and relevant graphene self-assembly
Cite this Article
Chen Qiao-Yue, Song Jun-Jie, Jing Liwei, Huang Kaikai, He Pimo, Zhang Hanjie. Triphenylene adsorption on Cu(111) and relevant graphene self-assembly. Chinese Physics B, 2020, 29(2): 026801 
Permissions
Triphenylene adsorption on Cu(111) and relevant graphene self-assembly
† Corresponding author. E-mail:
Project supported by the National Key Research and Development Program of China (Grant No. 2017YFB0503100) and the National Natural Science Foundation of China (Grant No. 11790313).
Abstract
Investigations on adsorption behavior of triphenylene (TP) and subsequent graphene self-assembly on Cu(111) were carried out mainly by using scanning tunneling microscopy (STM). At monolayer coverage, TP molecules formed a long-range ordered adsorption structure on Cu(111) with an uniform orientation. Graphene self-assembly on the Cu(111) substrate with TP molecules as precursor was achieved by annealing the sample, and a large-scale graphene overlayer was successfully captured after the sample annealing up to 1000 K. Three different Moiré patterns generated from relative rotational disorders between the graphene overlayer and the Cu(111) substrate were observed, one with 4° rotation between the graphene overlayer and the Cu(111) substrate with a periodicity of 2.93 nm, another with 7° rotation and 2.15 nm of the size of the Moiré supercell, and the third with 10° rotation with a periodicity of 1.35 nm.
1. Introduction
Graphene has far-ranging prospects in many fields, such as materials, energy, optics, and electricity, due to its excellent performance and far-reaching potential application values. The precondition of graphene application is to fabricate high quality, large scale continuous graphene. Graphene was firstly obtained by mechanical exfoliation from the highly oriented pyrolytic graphite.[1] Since then, various methods for preparing graphene have been developed, including chemical exfoliation of graphite,[2] epitaxial growth on SiC under ultra-high vacuum and high temperature,[3] chemical vapor deposition (CVD),[4,5] and molecular self-assembly of graphene.[6] The present researches upon molecular self-assembly to grow graphene mainly use aromatic hydrocarbon molecules as the precursor molecules and then perform annealing operation on the surface of transition metals. This process is easy to operate and graphene grown by this method has advantages of high quality. Furthermore, the width and edge geometry of graphene nanoribbons can be controlled by adjusting the annealing temperature and the coverage range of the precursor molecules.[7,8] For example, the width and edge shape of synthesized graphene nanoribbons on the Au(111) surface can be precisely controlled by molecular self-assembly method;[9] using C60 as the precursor molecule, uniform size graphene quantum dots can be grown on the Ru(0001) surface through this method.[10,11] However, for different substrates and precursor molecules, conditions for growing graphene are quite different, and there are few related public studies at present.[9,12,13]
Copper is one of the most commonly used substrate for synthesizing graphene. The principle of growing graphene on Cu substrate is very different from that on other substrates because the solubility of carbon in copper is very low, therefore, the movement of carbon rings on the copper surface can be simply considered as surface-based motion.[14–16] Graphene can be synthesized on the copper substrates with various crystal orientations, but the Cu(111) substrate is most widely used as it is easier to grow high-quality single-layer graphene. The interaction between graphene and the Cu(111) substrate is weak,[17,18] so different Moiré patterns generated from relative rotational disorders between graphene and the Cu(111) substrate can be observed. At present, researchers have mainly observed two different superstructures: the Moiré pattern of 0° rotation of the graphene lattice with the underlying Cu(111) substrate (∼ 6.6 nm periodicity),[16,19,20] and the Moiré pattern of 7° rotation of the graphene lattice with the underlying Cu(111) substrate (∼ 2 nm periodicity).[20–22] The Moiré pattern of 10.4° misorientation has also been found in recent experiments.[23]
In this paper, we chose triphenylene (C18H12, TP) as the precursor molecule to investigate the graphene self-assembly on the Cu(111) surface and analyzed Moiré patterns which formed with different misorientation angles. With scanning tunneling microscopy (STM), we first studied the growth and adsorption behavior of TP molecules on Cu(111) and then the processing of transforming TP molecules to graphene in the case of monolayer coverage. Finally, we analyzed the superstructures of different misorientation angles formed by aligning the graphene overlayer to the Cu(111) substrate rotationally.
2. Experimental and computational section
All the experiments were carried out in the ultra-high-vacuum variable temperature scanning probe system of Omicron, and the base pressure was better than 2 × 10−10 mbar. The system is consisted of a fast-entry lock, a sample preparation chamber, a sample analysis chamber, and an STM chamber.[24] The Cu(111) surface was cleaned by multiple cycles of argon ion sputtering (1000 eV for 1 hour) and subsequent high temperature annealing (at about 800 K). The cleanness and ordering of the sample were checked by low energy electron diffraction (LEED) and STM. Prior to experiment, TP was thoroughly degassed in a molecular beam epitaxy (MBE) chamber in a self-made tantalum boat. Before sublimation, the source was preheated under the heating power of 0.15 W. At the same time, the Cu(111) substrate remained unchanged at RT. During sublimation, the heating power was increased to 0.28 W, and maintained for 3 min. After 3 min of heating, the sample was directly opposed to the source for 10 s, and the growth rate of the film was about 0.1 ML/s. Afterwards the sample was directly passed to the analysis chamber for measurement. The pressure in the chamber was about 2 × 10−9 mbar during the film growth. STM measurements were conducted using constant current mode at RT. All the bias voltages were applied to the sample with respect to the tip. All experimental STM images in this paper were processed by WSxM software.[25]
3. Results and discussion
3.1. Adsorption of TP molecules on the Cu(111) surface
At sub-monolayer coverage, single TP molecule can not be imaged by STM at room temperature because of its high molecular mobility. This manifests the weak interaction between TP and Cu(111). For example, in order to visualize a single benzene ring on Cu(111) under low-coverage, both STM tip and substrate need to be putted in low temperature (∼ 5 K) during the experiment and when the temperature arrives at 77 K, the benzene ring starts to diffuse freely.[16,19,26,27] It has also shown that the interaction between aromatic hydrocarbon molecules and the Cu (111) substrate is always weak.[4–6,28–30] After ∼ 1 ML of TP deposition, the structure of TP molecules on Cu (111) substrate was successfully resolved at RT. Anyway, the adsorption energy of single TP molecule is higher than that of a benzene ring,[31] and a two-dimensional closely packed adsorption layer certainly enhanced the interaction between the molecules and substrate which minimized the influence of the tip–molecule interaction and the temperature. Previous investigations have already proven that planar TP molecules show a stable flat-lying orientation at a wide range of bias voltages from −0.5 V to −0.01 V.[32] Indeed, the increased bias voltage and tunneling current excite locally the molecules by a moderate electric field and tip–surface interaction.[33] Actually, in the present STM measurements, the absolute bias voltages below 0.6 V were applied to avoid destroying the formed monolayer ordering.
At monolayer coverage, TP molecules formed a long-range ordered adsorption structure on the Cu(111) surface with uniform orientation, which is detailedly figured out in Fig.
 | Fig. 1. (a) STM image with monolayer coverage of TP molecules on Cu(111) (50 nm × 50 nm, VT = −0.516 V, IT = 0.574 nA). (b) High resolution STM image (10 nm × 10 nm, VT = −0.447 V, IT = 0.088 nA). (c) Line profile along the straight line indicated in (b). (d) The enlarged STM image showing the molecular orientation in details. |
In order to infer the specific adsorption sites of TP on the Cu(111) surface, we first consider the possible adsorption sites of the single benzene ring on Cu(111) and its corresponding adsorption energy. Theoretical approach for benzene ring adsorption on Cu(111) shows that the three-fold hollow site is the most stable adsorption site (as shown in Fig.
 | Fig. 2. (a) The ball and stick model for benzene at hollow site of Cu(111), where the brown, gray, and white balls represent copper, carbon, and hydrogen atoms, respectively. (b) The model structure for TP on Cu(111) surface with p(5 × 5) supersymmetry. |
3.2. Graphene self-assembly on Cu(111) via TP molecules
In order to further investigate the conditions of graphene self-assembly on Cu(111) via TP molecules, the sample was first annealed at 600 K for 30 min. The catalytic influence of the substrate has been confirmed experimentally by the fact that cyclodehydrogenation on Cu(111) is already completed at 250 °C.[36] Therefore, annealing at 600 K ensured the cyclodehydrogenation of the TP precursor on the Cu(111) surface. The afterwards STM measurements showed that only seldom graphene fragments whose sizes were smaller than 3 nm × 3 nm were found at the step edges of the Cu(111) surface coexisting with products of polymerization (see Fig. S1 in
Finally, large-scale graphene films were successfully captured after sample annealing at 1000 K for 10 s, as shown in Fig.
 | Fig. 3. (a) STM image of graphene self-assembly on Cu(111) substrate via TP molecules after 1000 K annealing (100 nm × 100 nm, VT = −0.645 V, IT = 0.661 nA). The blue arrow refers to graphene boundaries. (b) Moiré pattern of graphene on Cu(111) at 7° misorientation angle (15 nm × 15 nm, VT = −0.987 V, IT = 0.282 nA), and the blue circle in the figure indicates defects found on the surface. Both the black arrows indicate the [110] direction of Cu(111). |
Based on the line profiles in STM images which are partly shown in Figs.
 | Fig. 4. (a) STM image of graphene coexistence with TP molecules (60 nm × 60 nm, VT = −0.530 V, IT = 0.972 nA). (b) Line profile along the yellow straight line outlined in Fig.  |
Density functional theory (DFT) and classical molecular dynamics simulations (CMD) are the most commonly used theoretical methods for studying the behavior of graphene on metal surfaces.[23,36,42] Recent researches have shown that the interaction between graphene and the Cu(111) substrate is van der Waals force based on DFT calculations.[35,41,43,44] Süle et al. developed a new Albell–Tersolf-like angular-dependent potential for the C–Cu interaction, but the results of periodicity of patterns simulated through this method are somewhat a bit lager than the STM measurements. CMD simulation showed that the adsorption energy has a minimum value (a stable phase) at a rotational angle of 10.4° between graphene and Cu(111) with a Moiré periodicity of 1.4 nm,[23] which agrees well with the value of 1.35 nm determined based on the STM images. The simple simulation by superimposing graphene lattice (0.246 nm) on Cu(111) (0.256 nm) with a 10° rotational angle (Fig. 
Figure 
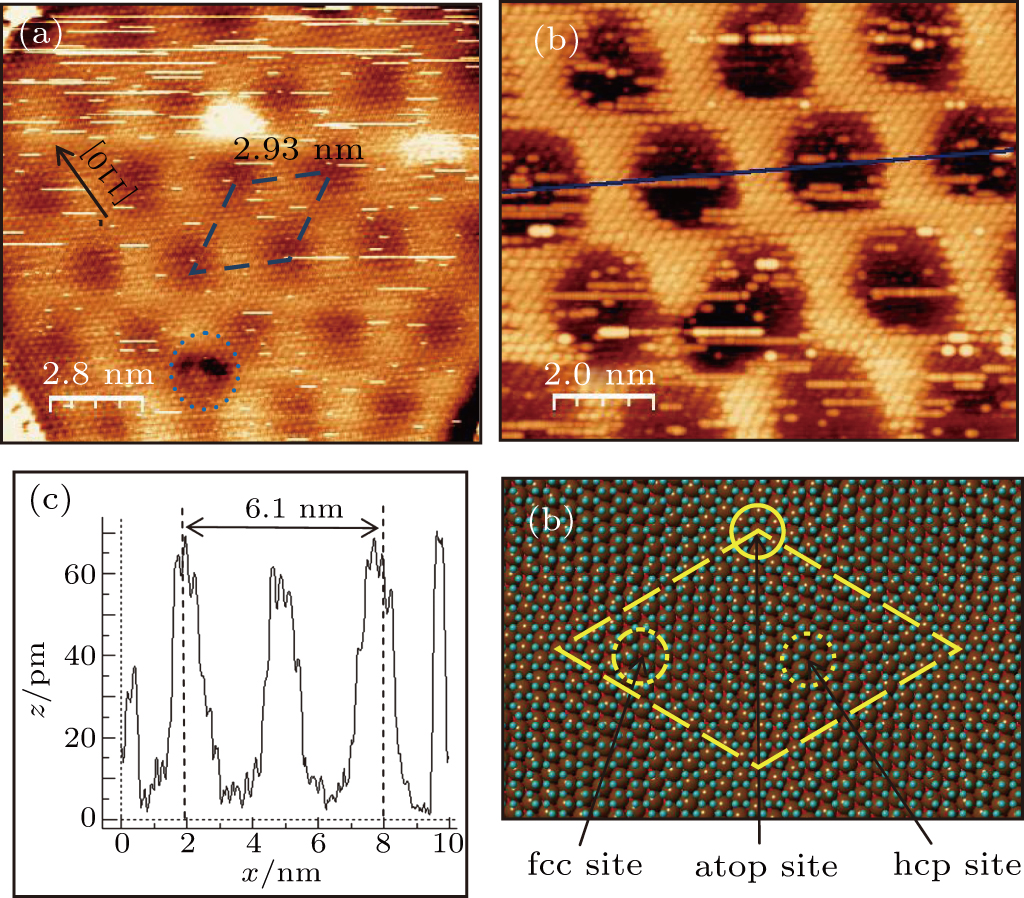 | Fig. 5. (a) STM topography of superstructure of graphene on Cu (111) with a periodicity of 2.93 nm (14 nm × 14 nm, VT = −0.172 V, IT = 0.558 nA), and the black arrow indicates the [110] direction of Cu(111). (b) High resolution STM topography (10 nm × 10 nm, VT = −0.183 V, IT = 0.403 nA). (c) Line profile along the blue straight line outlined in Fig.  |
4. Conclusions
In summary, we have investigated the growth of self-assembly graphene on the Cu(111) surface by using TP molecules as precursor and the Moiré patterns of graphene on Cu(111). At monolayer coverage, the weak balance between the molecule–molecule repulsive and molecule–substrate attractive interactions has favored 2D liquid-like growth which delicately formed a long-rang ordered superstructure. During sample annealing, at temperature ≤ 900 K, only small-scale graphene fragments can form. But after annealing at higher temperature (1000 K), the dissociation of TP molecules and small graphene dissociation, removal of Cu oxide, and increasing mobility of graphene islands contribute to forming the large-scale graphene. Three different Moiré patterns generated from relative rotational disorders between graphene and the Cu(111) substrate have been observed and according to the result inferred from DFT and CMD, we map each pattern to its corresponding rotation: one with 4° rotation between graphene lattice and the Cu(111) substrate with the periodicity of 2.93 nm which has not been found in other research, another with 7° rotation and the size of the Moiré supercell is 2.15 nm, and the third with 10° rotation with a periodicity of 1.35 nm.
Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] | |
| [33] | |
| [34] | |
| [35] | |
| [36] | |
| [37] | |
| [38] | |
| [39] | |
| [40] | |
| [41] | |
| [42] | |
| [43] | |
| [44] | |
| [45] | |
| [46] | |
| [47] | |
| [48] |