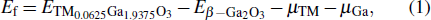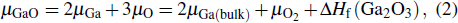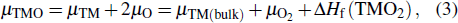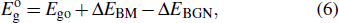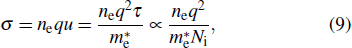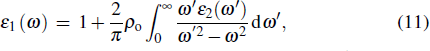1. Introductionβ-Ga2O3 is an emerging semiconductor material with a wide bandgap of about 4.9 eV. β-Ga2O3 exhibits fascinating physical properties and chemical stability, such as good transparency and a critical breakdown field of up to 8 MV/cm, which has made it a focus of attention in recent years.[1,2] With these advantages, β-Ga2O3 is considered to be a promising transparent conducting material operating from the visible to the deep-ultraviolet spectrum for photodetector and other optoelectronic devices.[3,4]
However, the poor electrical conductivity of β-Ga2O3 hinders its application as a transparent conducting oxide (TCO).[5] It is well known that doping is an effective way to improve the photoelectric properties of materials, including conductivity, mobility and absorptivity. Si is regarded as a suitable dopant for n-type β-Ga2O3 because excess free electrons will be generated after doping. Further, the oxides of Si and Ga have a similar melting point, which is beneficial for adjusting the growth temperature and obtaining a high doping concentration. Zhang et al.[6] obtained a Si-doped β-Ga2O3 film using the pulsed laser deposition (PLD) technique, and the highest doping concentration is 9.1 × 1019 cm−3, but the corresponding conductivity is only 2.0 S · cm−1. The low conductivity is due to the amorphous-like structure of the Si-doped β-Ga2O3 and blockage of electronic transmission paths. Takakura et al.[7] deposited Si-doped β-Ga2O3 film by radio-frequency (RF) magnetron sputtering, and found that the optical gap of the Si-doped β-Ga2O3 is wider than that of β-Ga2O3. Recently, a homoepitaxial Si-doped Ga2O3 film produced by PLD, with a high carrier concentration of 1.74 × 1020 cm−3 and ultrahigh conductivity of 732 S/cm, was reported,[8] but its conductivity is still far below that of commercial ITO film or Al-doped ZnO film. Sn is another promising dopant as a donor for improving the conductivity of β-Ga2O3 because excess free electrons will be generated when Sn4+ substitutes Ga3+. Orita et al.[1] deposited n-type Ga2O3 film by Sn doping and obtained the best conductivity of 8.2 S · cm−1. Ohira et al.[5] prepared Sn-doped β-Ga2O3 by the float-zone method, found that Sn was dispersed in β-Ga2O3 uniformly, and suggested that Sn be substituted in the Ga site in the β-Ga2O3 lattice, but they did not discuss the optical properties of Sn-doped β-Ga2O3. Baldini et al.[9] grew Si- and Sn-doped homoepitaxial β-Ga2O3 films by metal organic vapor phase epitaxy (MOVPE), and the highest electron concentrations of 8 × 1019 cm−3 for Si-doped β-Ga2O3 and 1 × 1019 cm−3 for Sn-doped β-Ga2O3 were obtained. The corresponding electron mobilities were 54.37 cm2/V · s and 32 cm2/V · s, but the conductivities and optical properties were not analyzed.
First-principles calculations based on density functional theory (DFT) have been extensively used for scientific research, and some material properties have been successfully parsed, such as electronic structures, optics and magnetism. Varley et al.[10] found that a stable structure is obtained when Si4+ replaces Ga3+ in the four-fold coordination according to the calculated total energy of Si-doped Ga2O3 by using first principles. Siah et al.[11] reported that Sn4+ prefers six-fold coordination and tends to substitute the Ga3+ octahedral site for Sn-doped β-Ga2O3. Zhang et al.[12] calculated the electronic structures and optical properties of β-Ga2O3 and Sn-doped β-Ga2O3 by using the generalized gradient approximation (GGA) exchange–correlation function based on DFT, but the calculated bandgap of β-Ga2O3 is only 2.315 eV, which is less than half of the experimental value (4.9 eV[1]). The underestimation of the bandgap is a well-known drawback of the standard DFT calculation due to the lack of precise consideration of the exchange–correlation potential, but it can be corrected by the scissors approximation,[13] hybrid-functional,[14] and GGA + U methods.[15] In order to obtain calculated bandgaps that are close to the experimental data, Li et al.[14] calculated the electrical and optical properties of β-Ga2O3 and Si-doped β-Ga2O3 using the screened hybrid functional HSE06, but the calculated result shows that β-Ga2O3 is a direct-bandgap material, which is not consistent with the experimental result.[16] Lately, Dong et al.[15] calculated the influences of oxygen vacancies on the structural and optical properties of β-Ga2O3 by GGA + U approximation with the Hubbard U parameters of 7.0 eV and 8.5 eV for Ga and O ions, respectively. The calculated bandgap of β-Ga2O3 is 4.92 eV, which is in agreement with the experimental value, but the calculated peak of the Ga 3d state is located at about −14.8 eV, which deviates from the experimental result of −17.12 eV,[17] and so the Hubbard U value requires reconsideration. So far, there have been no reports on the electronic structures and optical properties of Si-doped and Sn-doped β-Ga2O3 by using GGA/LDA + U methods. There are also very few comparative research reports on the physical properties of Si-doped β-Ga2O3 and Sn-doped β-Ga2O3. To further understand the differences in the electronic structures and optical properties of Si-doped β-Ga2O3 and Sn-doped β-Ga2O3, the energy band structures, density of states and optical properties are calculated in this paper using the GGA + U method based on DFT.
3. Results and discussion3.1. Structural propertiesThe optimized structure parameters of β-Ga2O3 and TM-doped β-Ga2O3 (TM = Si,Sn) are listed in Table 1. It is clear that the calculated lattice constants of β-Ga2O3 are in good agreement with the experimental results[37] and other calculated data,[38] which indicates that the calculation method and models are rational. The lattice constants of β-Si0.0625Ga1.9375O3 and β-Sn0.0625Ga1.9375O3 are larger than those of β-Ga2O3, which mainly originates from the competitive effects of the Coulomb repulsion and the ionic radius difference between Si and Ga or Sn and Ga. On the one hand, the excess free electrons will be generated and will enhance the Coulomb repulsion when Ga3+ is replaced by Si4+ or Sn4+, and the strong effect of the Coulomb repulsion will increase the supercell volume. On the other hand, the larger ionic radius will increase the volume of the materials. For β-Si0.0625Ga1.9375O3, the Si ionic radius (0.026 nm[27]) is much smaller than that of the Ga(1) (0.047 nm[28]), but the effect of the Coulomb repulsion plays a more important role than the ionic radius difference, so the result of the competition is the supercell volume of β-Si0.0625Ga1.9375O3 being larger than that of β-Ga2O3. For β-Sn0.0625Ga1.9375O3, the Sn ionic radius (0.069 nm[29]) is larger than that of the Ga(2) (0.062 nm[28]), and therefore the total superposition makes the supercell volume of β-Sn0.0625Ga1.9375O3 larger than that of β-Ga2O3.
Table 1.
Table 1.
 Table 1. The structural parameters and formation energies of β-Ga2O3 and TM-doped β-Ga2O3 (TM = Si,Sn) under the GGA + U method. .
|
|
Lattice constant |
Volume/Å |
Ef/eV |
|
|
a/Å |
b/Å |
c/Å |
O-poor |
O-rich |
| β-Ga2O3 |
this work |
12.494 |
3.092 |
5.900 |
221.949 |
|
|
| Exp.[25] |
12.220 |
3.038 |
5.786 |
|
|
|
| Calc.[26] |
12.438 |
3.084 |
5.877 |
|
|
|
| β-Si0.0625Ga1.9375O3 |
|
12.516 |
3.093 |
5.907 |
221.998 |
1.798 |
4.291 |
| β-Sn0.0625Ga1.9375O3 |
|
12.561 |
3.111 |
5.926 |
225.007 |
1.120 |
3.615 |
| Table 1. The structural parameters and formation energies of β-Ga2O3 and TM-doped β-Ga2O3 (TM = Si,Sn) under the GGA + U method. . |
3.2. Doping formation energyTo evaluate the structure stability of TM-doped β-Ga2O3 (TM = Si,Sn), the doping formation energies of β-Si0.0625Ga1.9375O3 and β-Sn0.0625Ga1.9375O3 are calculated according to the formula[30]
where
ETM0.0625Ga1.9375O3 is the total supercell energy of
β-Si
0.0625Ga
1.9375O
3 or
β-Sn
0.0625Ga
1.9375O
3,
Eβ−Ga2O3 is the total supercell energy of
β-Ga
2O
3,
μTM represents the chemical potential of the Si or Sn atom, and
μGa is the chemical potential of the Ga atom. The chemical potential is connected with the growth atmosphere of the materials, which further affects the doping formation energies of TM-doped
β-Ga
2O
3. Therefore, the doping formation energies of
β-Si
0.0625Ga
1.9375O
3 and
β-Sn
0.0625Ga
1.9375O
3 are calculated under O-rich and O-poor conditions. To determine the values of
μGa and
μTM, the formation enthalpies Δ
Hf of
β-Ga
2O
3, SnO
2, and SiO
2 are considered assuming their formations from the metallic Ga, Sn, semiconductive Si, and gaseous O
2. The chemical potentials of
μGa and
μTM are calculated using the following relationships:
where
μGaO and
μTMO are the energies of
β-Ga
2O
3 and TMO
2 in a single molecule, and
μGa(bulk),
μTM(bulk), and
μO2 are the chemical potentials of metallic Ga, Sn, semiconductive Si, and gaseous O
2, respectively. By combining Eqs. (
2) and (
3), the chemical potentials of Ga and TM are
for O-poor conditions, and
for O-rich conditions. The calculated formation enthalpy Δ
Hf(Ga
2O
3) for
β-Ga
2O
3 is −10.95 eV, which is close to the experimental value of −11.3 eV.
[10] The doping formation energies of
β-Si
0.0625Ga
1.9375O
3 or
β-Sn
0.0625Ga
1.9375O
3 under O-poor and O-rich conditions are calculated using Eqs. (
4) and (
5), and the calculated values are listed in Table
1. As can be seen in Table
1, the doping formation energies of
β-Si
0.0625Ga
1.9375O
3 or
β-Sn
0.0625Ga
1.9375O
3 under O-poor conditions are lower than those under O-rich conditions, which indicates that the TM-doped
β-Ga
2O
3 (TM = Si,Sn) tend to form under O-poor conditions. Moreover, the doping formation energy of
β-Si
0.0625Ga
1.9375O
3 is larger than that of
β-Sn
0.0625Ga
1.9375O
3 whether under O-rich or O-poor conditions, which reveals that the preparation of Si-doped
β-Ga
2O
3 requires a higher growth temperature as compared to Sn-doped
β-Ga
2O
3. To explain the difference in the doping formation energy between
β-Si0
.0625Ga
1.9375O
3 and
β-Sn0
.0625Ga
1.9375O
3, the average bond lengths of Ga(1)–O, Si–O, Ga(2)–O, and Sn–O are given in Table
2. It is well known that the bond length variation is related to the deformation and relaxation of the material structure, which is reflected in the formation energy. It is found from Table
2 that the bond length variation between Ga(1)–O and Si–O is significantly larger than that between Ga(2)–O and Sn–O. Larger bond length variation requires more energy, so the doping formation energy of
β-Si
0.0625Ga
1.9375O
3 is larger than that of
β-Sn
0.0625Ga
1.9375O
3.
Table 2.
Table 2.
Table 2. The average bond lengths of Ga–O, Si–O and Sn–O for TM-doped β-Ga2O3 (TM = Si,Sn). .
|
Ga(1)–O |
Si–O |
Ga(2)–O |
Sn–O |
| Average/Å |
1.875 |
1.665 |
2.033 |
2.129 |
| Variation/% |
11.2 |
4.5 |
| Table 2. The average bond lengths of Ga–O, Si–O and Sn–O for TM-doped β-Ga2O3 (TM = Si,Sn). . |
3.3. Energy band structure and density of statesTo illustrate the effect of the Hubbard U on the band structure and density of states, the calculated band structures and densities of states (DOSs) of β-Ga2O3 under GGA and GGA + U methods are shown in Fig. 2. The high symmetry points in the Brillouin zone are located at Γ(0,0,0), A(0,0.5,0), H(0,0.5,0.5) and K(0,0,0.5), and the Fermi level is defined as 0 eV. It can be seen in Fig. 2 that β-Ga2O3 has an indirect bandgap with the conduction band minimum (CBM) at Γ point and the valence band maximum (VBM) at M point, which is in agreement with the experimental results.[16] But the direct Γ+−Γ− transition is dominant in the optical spectra because the difference between the direct bandgap (Γ+–Γ−) and the indirect bandgap (Γ+–M) is very small (about 0.023 eV), which is proved by the analysis of the dipole transition matrix element and the experimental results of transmission spectra,[31] so the direct Γ+–Γ− transition of β-Ga2O3 merits more attention than the indirect Γ+–M transition in optics. As can be seen in Fig. 2(a), the bandgap of β-Ga2O3 under the GGA approximation is only 1.949 eV, which deviates significantly from the experimental result of 4.9 eV.[1] Besides, the upper valence bandwidth is only 6.78 eV, which does not agree with the experimental value of about 7.37 eV.[16] More importantly, the Ga 3d state peak is located at 11.63 eV below the VBM, which is not consistent with the experimental result of −17.12 eV.[17] In order to correct the above deviation, the GGA + U method is used and the corrected bandgap of β-Ga2O3 is about 4.886 eV (see Fig. 2(b)). At the same time, the upper valence bandwidth is 7.29 eV and the Ga 3d state peak is at −17.01 eV, which are in good agreement with the previous experimental results, so the electronic structures calculated by the GGA + U method are reasonable. Moreover, as can be observed from the partial DOSs in Fig. 2(b), the CBM is dominated by the Ga 4s state and the VBM is occupied by the O 2p state, so the bandgap of β-Ga2O3 is determined by O 2p and Ga 4s states, which is consistent with the other calculations.[32]
To further investigate the effect of the doping atom on the electronic structure of β-Ga2O3, the band structures and DOSs of β-Si0.0625Ga1.9375O3 and β-Sn0.0625Ga1.9375O3 obtained by the GGA + U method are shown in Fig. 3. The bandgaps Eg of β-Si0.0625Ga1.9375O3 and β-Sn0.0625Ga1.9375O3 are 4.821 eV and 4.613 eV, which are smaller than that of β-Ga2O3. But the optical gaps
 (defined as the vertical distance from the Fermi level to the VBM) of β-Si0.0625Ga1.9375O3 and β-Sn0.0625Ga1.9375O3 are 5.853 eV and 5.586 eV, larger than that of β-Ga2O3, which is consistent with the experimental result.[7] The electron transition of absorption spectra is related to the optical gap, and the optical gap is an important physical parameter for a TCO material, so β-Si0.0625Ga1.9375O3 has the largest optical gap and is more suitable for application in photoelectric devices as a deep-ultraviolet TCO than are β-Ga2O3 and β-Sn0.0625Ga1.9375O3.
(defined as the vertical distance from the Fermi level to the VBM) of β-Si0.0625Ga1.9375O3 and β-Sn0.0625Ga1.9375O3 are 5.853 eV and 5.586 eV, larger than that of β-Ga2O3, which is consistent with the experimental result.[7] The electron transition of absorption spectra is related to the optical gap, and the optical gap is an important physical parameter for a TCO material, so β-Si0.0625Ga1.9375O3 has the largest optical gap and is more suitable for application in photoelectric devices as a deep-ultraviolet TCO than are β-Ga2O3 and β-Sn0.0625Ga1.9375O3.
The total and the partial densities of states of β-Si0.0625Ga1.9375O3 and β-Sn0.0625Ga1.9375O3 are also given in Fig. 3. For β-Si0.0625Ga1.9375O3, the VBM is occupied by the O 2p state, whereas the CBM is occupied by the Si 3s state, so the bandgap of β-Si0.0625Ga1.9375O3 is determined by O 2p and Si 3s states. It can also be seen in Fig. 3(a) that the DOS of β-Si0.0625Ga1.9375O3 moves along the low energy direction after doping and the Fermi level enters into the conduction band, which indicates the n-type semiconductor feature. Therefore, the conductivity of Si-doped β-Ga2O3 is significantly improved compared to that of β-Ga2O3, which is consistent with the experiment result.[33] For β-Sn0.0625Ga1.9375O3 in Fig. 4(b), the VBM is occupied by the O 2p state and the CBM is occupied by the Sn 5s state, so the bandgap of β-Sn0.0625Ga1.9375O3 is determined by O 2p and Sn 5s states. Further, the free electrons from Sn 5s state fill the CBM and improve the conductivity of β-Sn0.0625Ga1.9375O3, which is similar to the scenario for β-Si0.0625Ga1.9375O3.
3.4. Electron effective mass and conductivityFor a doped semiconductor with high doping concentration (i.e., >1019 cm−3), there are primarily two competing mechanisms affecting its optical gap: the Burstein–Moss effect[34] and the bandgap renormalization effect.[35] The Burstein–Moss effect causes bandgap widening due to the Fermi-band filling effect, marked as ΔEBM. The bandgap renormalization effect causes bandgap narrowing due to the many-body bandgap renormalization effect and the semiconductor–metal transition effect, labeled as ΔEBGN. The ΔEBM and ΔEBGN can be obtained by[36]
where
Ego denotes the bandgap of the undoped
β-Ga
2O
3,
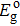
is the optical gap, and
Eg is the bandgap. According to the band structures in Figs.
2–
4,
Ego is 4.886 eV, and
Eg is 4.821 eV for
β-Si
0.0625Ga
1.9375O
3 and 4.613 eV for
β-Sn
0.0625Ga
1.9375O
3. By Eqs. (
6) and (
7), the Δ
EBM of
β-Si
0.0625Ga
1.9375O
3 and
β-Sn
0.0625Ga
1.9375O
3 are 1.032 eV and 0.973 eV, and the Δ
EBGN of
β-Si
0.0625Ga
1.9375O
3 and
β-Sn
0.0625Ga
1.9375O
3 are 0.065 eV and 0.273 eV, respectively. Further, the electron effective mass
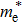
is related to Δ
EBM by the following formula:
[12]
where
h is the Planck constant, and
ne is the electron concentration. Using Eq. (
8), the electron effective mass of
β-Si
0.0625Ga
1.9375O
3 is 0.383
me (
me is the electron mass) and 0.402
me for
β-Sn
0.0625Ga
1.9375O
3. The electron conductivity of the n-type material can be expressed as
[37]
where
q is the electron charge,
μ is the electron mobility and
τ is the relaxation time which is related to the ionized concentration (
Ni) and thermodynamic temperature. According to Eq. (
9), the electron mobility ratio of
β-Si
0.0625Ga
1.9375O
3 to
β-Sn
0.0625Ga
1.9375O
3 is 1.035, revealing that the electron mobility of
β-Si
0.0625Ga
1.9375O
3 is higher than that of
β-Sn
0.0625Ga
1.9375O
3, which is consistent with recent experimental results.
[9] Further, the electron conductivity ratio of
β-Si
0.0625Ga
1.9375O
3 to
β-Sn
0.0625Ga
1.9375O
3 is 1.065 from Eq. (
9), so Si-doped
β-Ga
2O
3 has an advantage over Sn-doped
β-Ga
2O
3 in terms of conductivity.
3.5. Optical propertiesOptical properties are important for TCO materials and are expressed by the complex dielectric function ε(ω) = ε1(ω) + iε2(ω). The imaginary part ε2(ω) of the complex dielectric function is determined by the momentum matrix elements between the occupied and unoccupied wave functions. The real part ε1(ω) can be evaluated from imaginary part ε2(ω) using the Kramers–Kronig relationship, and the absorption coefficient α(ω) can be calculated according to the complex dielectric function. The above relationships are described by the following formulas:[38]
Here,
ρo represents the principal value of the integral, subscripts C and V represent the conduction band and valence band states, respectively, BZ means the first Brillouin zone,
k is the reciprocal lattice vector, |
MCV(
k)|
2 denotes the momentum matrix element,
c is a constant,
ω stands for the angular frequency, and
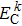
and
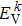
represent the intrinsic energy levels. The imaginary part
ε2(
ω) of the complex dielectric function is the basis for analyzing the optical properties of any materials.
The imaginary parts ε2(ω) of the complex dielectric function of β-Ga2O3, β-Si0.0625Ga1.9375O3 and β-Sn0.0625Ga1.9375O3 in the x, y, and z directions are calculated and shown in Fig. 4. For the ε2(ω) of β-Ga2O3, three main peaks at 9.286 eV, 10.587 eV, and 9.506 eV appear in the x, y, and z directions; the corresponding peak intensities are 1.42, 1.316, and 2.134, respectively, which indicates that β-Ga2O3 has significant anisotropy. For Si- and Sn-doped β-Ga2O3, there are no significant changes in the position of the three main peaks in the x, y, and z directions, but in the low-energy range, two small peaks (about 0.9 eV for β-Sn0.0625Ga1.9375O3 and about 1.05 eV for β-Si0.0625Ga1.9375O3) appear. It is well known that the imaginary part ε2(ω) is related to the electron transition between the energy bands, so the peak of ε2(ω) can be explained by the electronic structures. According to the partial DOS in Fig. 3, the main peak of about 10 eV mainly originates from the interband electron transition between the Ga 4s state of the conduction band and the O 2p state of the valence band, but the main peak edges (near to 5 eV) of β-Ga2O3, β-Si0.0625Ga1.9375O3 and β-Sn0.0625Ga1.9375O3 are different because their interband electron transitions are different (the electron transitions of Ga 4s and O 2p for β-Ga2O3, Si 3s and O 2p for β-Si0.0625Ga1.9375O3, and Sn 5s and O 2p for β-Sn0.0625Ga1.9375O3). The small peak at about 0.9 eV for β-Sn0.0625Ga1.9375O3 is mainly derived from the electron transition between the Sn 5s state and the O 2p state in the conduction band, and the small peak at about 1.05 eV for β-Si0.0625Ga1.9375O3 is mainly derived from the electron transition between the Si 3s state and the O 2p state in the conduction band. The strength difference of the two small peaks is mainly attributed to the different bonding structures: a Si atom bonds with the four closest oxygen atoms but a Sn atom bonds with the six closest oxygen atoms.
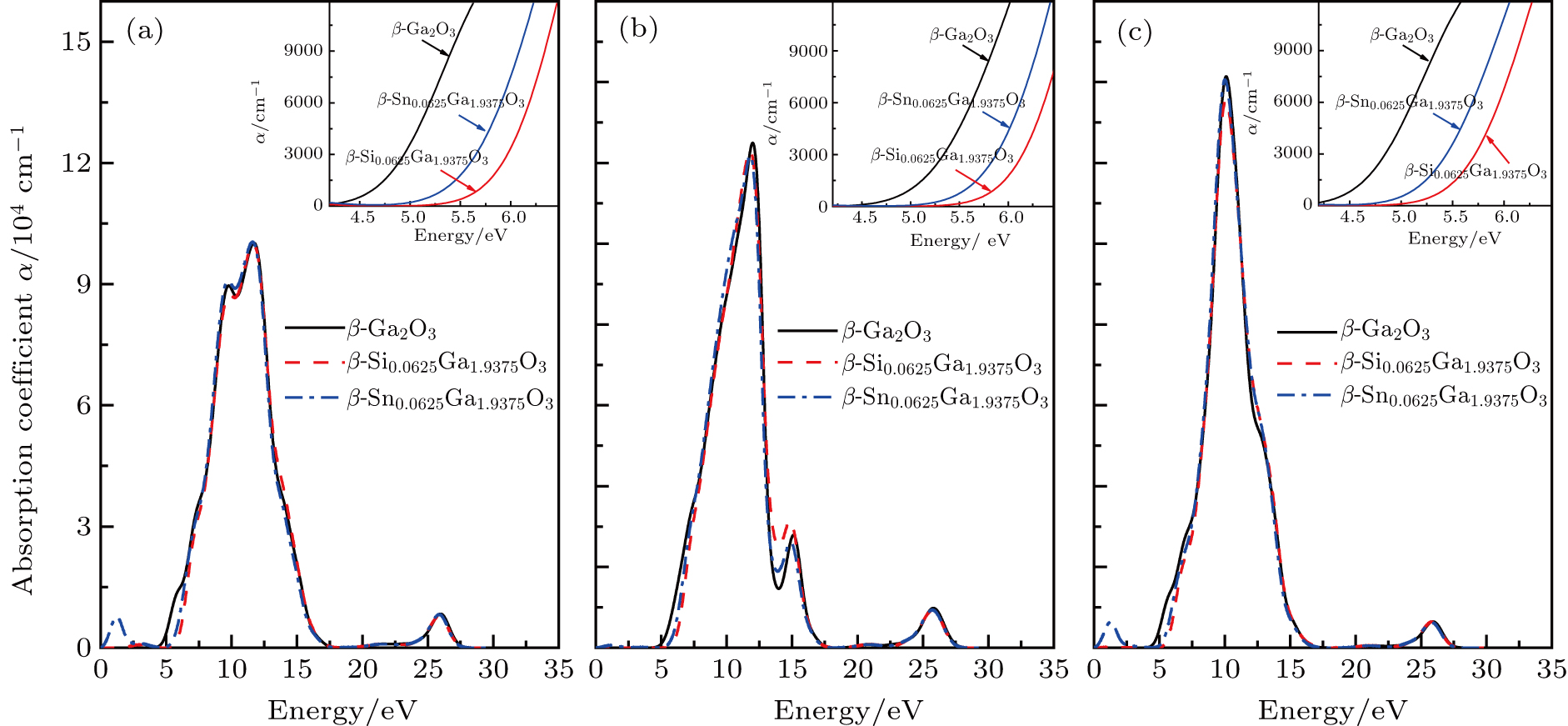
The absorption coefficients α(ω) of β-Ga2O3, β-Si0.0625Ga1.9375O3 and β-Sn0.0625Ga1.9375O3 in the x, y and z directions are plotted in Fig. 5, and the insets are the partial enlargement. As shown in Fig. 5, the absorption spectra in the x-, y- and z-axis directions are mainly distributed between 5 eV and 17 eV, which are consistent with the imaginary parts ε2(ω) of the complex dielectric function. The main peaks of the absorption spectra along the x-, y- and z-axes are located at 11.6 eV, 12.04 eV and 10.03 eV, respectively, and the peak intensity gradually increases, which is slightly different from the main peaks of the imaginary parts ε2(ω) due to the effect of the real part ε1(ω) of the complex dielectric function. The main peak shapes and intensities are different in the x, y and z directions due to the anisotropy structure, but the peak shapes of β-Ga2O3, β-Si0.0625Ga1.9375O3 and β-Sn0.0625Ga1.9375O3 in the same direction are hardly changed except for the edge of the main peak, which means that the main peak is derived from the same electron transition of the Ga 4s and O 2p states and the edge of the peak is related to Si or Sn doping.
To analyze clearly the difference in the peak edge, the spectra between 4.2 eV and 7 eV are also given in the insets of Fig. 5. It can be observed that the optical absorption edges of β-Ga2O3 in the x, y, and z directions are 4.753 eV, 5.152 eV and 4.646 eV, respectively, and this explains the cause of the different experimental bandgaps (4.6–5.1 eV) of β-Ga2O3 in the literature.[39,40] Moreover, the average optical absorption edge of β-Ga2O3 is 4.85 eV, which is very close to the bandgap (4.886 eV) of β-Ga2O3 from the band structure in Fig. 2. For β-Si0.0625Ga1.9375O3, the optical absorption edges in the x, y and z directions are 5.897 eV, 6.017 eV and 5.64 eV, respectively, and the average optical absorption edge is 5.851 eV, close to the optical gap of 5.853 eV in Fig. 3. For β-Sn0.0625Ga1.9375O3, the optical absorption edges in the x, y and z directions are 5.592 eV, 5.813 eV and 5.282 eV, respectively, and the average optical absorption edge is 5.562 eV, which is consistent with the optical gap (5.586 eV) of β-Sn0.0625Ga1.9375O3, so the optical absorption edge can be described as a joint band structure between the Fermi level and the VBM. Further, it is noted that the optical absorption edge in the y direction is the largest among the three directions, and the average optical absorption edge of β-Si0.0625Ga1.9375O3 is the largest among them. Figure 5 shows that two small absorption peaks of about 1.2 eV in the x and z directions occur in β-Sn0.0625Ga1.9375O3, but no obvious absorption peaks are found in β-Ga2O3 and β-Si0.0625Ga1.9375O3. Therefore, β-Si0.0625Ga1.9375O3 has better light transmittance in the deep-ultraviolet wavelength range than do β-Ga2O3 and β-Sn0.0625Ga1.9375O3.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}