



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Tracking coherent low frequency vibrational information of Rh101 in ground and excited electronic states by broadband transient grating spectroscopy
Cite this Article
Zhang Wei, Liu Xiao-Song, Wang Zan-Hao, Song Yun-Fei, Yang Yan-Qiang. Tracking coherent low frequency vibrational information of Rh101 in ground and excited electronic states by broadband transient grating spectroscopy. Chinese Physics B, 2018, 27(12): 123301 
Permissions
Tracking coherent low frequency vibrational information of Rh101 in ground and excited electronic states by broadband transient grating spectroscopy
† Corresponding author. E-mail:
Project supported by the Science Challenge Project, China (Grant No. TZ2016001) and the National Natural Science Foundation of China (Grant No. 21673211).
Abstract
Time- and frequency-resolved broadband transient grating (BB-TG) spectroscopy is used to distinguish between ground- and excite-electronic state vibrational coherence at different wavelengths. Qualitative theoretical analysis using double-sided Feynman diagrams indicates that a superposition of ground and excited state vibrational coherence are contained in the ground state absorption (GSA) and stimulated emission (SE) overlap band, while only the excited state is contained in the excited state absorption (ESA) band. The TG experiment, in which a white light continuum (WLC) is adopted as a probe, is conducted with rhodamine101 (Rh101+) as the target molecule. Fourier analysis of TG dynamics in a positive delay time range at specific wavelengths enables us to distinguish the low-frequency vibrational modes of Rh101 in ground- and excite-electronic states.
1. Introduction
At a molecular level, the coherence and relaxation of vibrations play an important role in chemical reactions, both in the electronic ground state and in the excited state (which are named ground state and excited state hereafter). The research on the vibrational coherence of specific molecules is helpful to enhance the cognition of vibrational evolution during electronic transition, tracing the chemical reaction path,[1] and even designing and manufacturing photo-switches, and to some extent molecular machines.[2–4]
A direct method to obtain the vibrational information of a given molecule in different electronic states is to measure its frequency domain; i.e., off-resonant combination with resonant Raman technique.[5] However, low-frequency vibrational modes (< 100 cm−1) are difficult to observe in the frequency domain in liquid ambient due to the intense Rayleigh wing. Furthermore, like laser dyes, the electronic excited state vibrations of a molecule are very difficult to distinguish in the frequency domain due to their strong fluorescence.
In recent years, with the advent of a femtosecond laser[6] whose pulse duration is shorter than plentiful photochemical and photophysical processes, the evolution of specific vibrational wavepackets in ground and electronic excited states can be traced and monitored in the time domain. The molecular vibrational modes, even with low frequency, can be obtained via Fourier transform (FT). In addition, the time domain measurement is immune to the fluorescence noise in excited states. A large number of studies[7–13] have proven its feasibility and two main methods have been established to distinguish vibrational coherence from different electronic states. The first method is Fourier analysis for different time periods of the dynamics, to be specific, positive (probe pulse goes after pump pulse in time) and negative (probe preceding pump pulse) periods. Kiefer and coworkers[9] used iodine vapor as a model molecule and studied the dynamics in different time ranges by using coherent anti-Stokes Raman scattering (CARS) technique with the pump and probe pulses resonant with the sample. Their results indicated that for the negative range, the CARS transient revealed the dynamics of the excited state of iodine; while for positive delay time, both the ground state and excited state dynamics were probed.[7,9] In addition, coherent Stokes Raman scattering (CSRS),[9] four-wave mixing (FWM)[9,12] and transient grating (TG)[8,14] technique were also discussed to separate electronic ground state vibrational dynamics in the same way as the excited state. To obtain a more precise excited state vibrational frequency, a relatively long period of transient in negative delay time range is needed. Meanwhile, the broadband transient absorption (TA) technique has also been employed to study the dynamics and vibrational coherence of different molecules. The dynamics of an excited state can be distinguished from ground state similarly.[10,11]
The other method is broadband impulsive vibrational spectroscopy (BB-IVS), which was established by Kukura et al.[13] Their approach was based on impulsive stimulated Raman scattering (ISRS),[15] and simply used a sufficiently short laser pulse whose time duration is shorter than the vibration period to exert an impulsive driving force to coherently stimulate corresponding vibration modes.[16] They combined the off-resonant ultrashort impulsive pulse with a time delayed uncompressed white light continuum (WLC) probe to record ground state vibrational coherences in the time domain.[17] If there is an actinic laser that is resonant with the sample molecule stimulated mixture of ground- and excited-state molecules before the impulsive pulse, then both ground- and excited-state vibrational coherence can be traced.[18] However, the excited-state vibrational modes are mixed up with those in the ground state, which makes it difficult to distinguish the vibrational information from different states.
Although both of these methods can detect vibrational coherences in different electronic states, each has limitations. In particular, the vibrational frequency obtained by first method is of low accuracy due to the short electronic dephasing time.[10] While for the second method, at least two experiments are needed to distinguish the vibration information from different electronic states.
In the present research, the BB-TG technique is adopted to detect the vibrational coherence in different electronic states. The direction of TG signal is separated from the direction of the pump and probe light, so the technique can realize ‘back-ground free detection,’ which gives rise to an extremely high signal-to-noise ratio. A WLC is used as the probe light, which can cover broadband spectral range, so the dynamic properties of different electronic states can be detected at different wavelengths. In virtue of broadband frequency coverage and accurate time resolution, only one measurement in positive delay time is needed to differentiate the vibrational coherence of ground-state from that of excited-state. Compared with the previously mentioned methods, the BB-TG technique provides a concise and effective way to investigate the vibrational information of specific molecules.
2. Theoretical background
where P (3)(k s, t) represents the time-dependent third-order polarization under a specific phase matching condition. This equation is applicable for the homodyne detection. For the TG measurements in this research, P (3)(k s, t) can be defined as[20,21]
where χ (3)(t) is the third order susceptibility associated with the system, E a and E b are the electric field of the pulses forming the TG and the electric field that scatters from the TG, respectively,
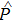
represents the polarization, and
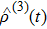
denotes the third-order density operator of the signal.
The TG is an FWM technique that is often employed to study molecular dynamics. The time sequence and beam configuration of pump and probe light are exhibited in Figs.
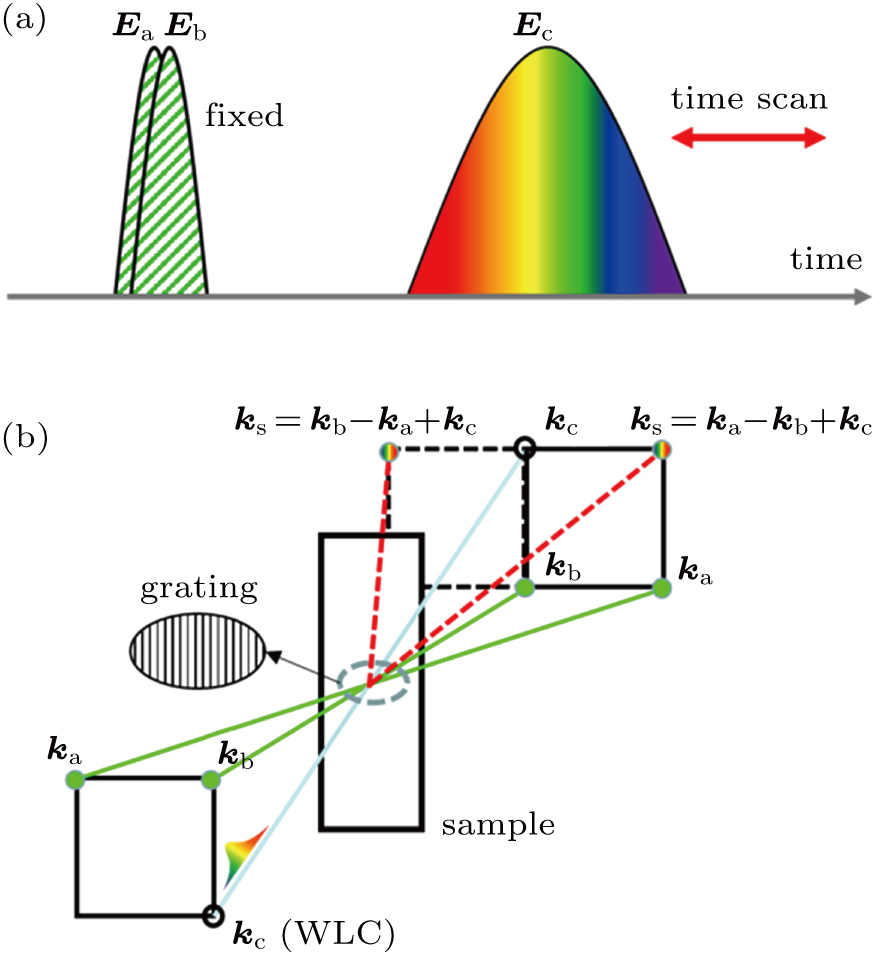 | Fig. 1. (color online) (a) Time sequence of the pump and probe pulse, where two pump pulses are fixed in time while probe is of time scanning. (b) Beam configuration of pump, probe, and TG signal in TG measurement. |
There are several beam configurations that can be adopted to realize TG measurement. In this study, forward folded BOXCARS[19] arrangement is chosen, as shown in Fig.
The TG signal intensity (ITG) that results from the interaction between three incident laser pulses and sample medium can be estimated from[20]
 |
 |
The evolution of the system under the influence of the incident electromagnetic field and the intrinsic relaxation process can be described by 

In this research, GSA, SE, and ESA bands in positive delay time range of TG measurement were all considered. Six possible diagrams were involved to describe the TG signal under RWA: four are applicable to SE & GSA overlapping band while the other two hold true in ESA process. The four diagrams shown in Figs. 



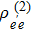

 | Fig. 2. Double-sided Feynman diagrams corresponding to (a) GSA, (b) SE, and (c) ESA of TG in positive delay time range, where Ea and Eb denote pump pulses while Ec presents probe pulse. |
The homologous diagrams for the ESA band are shown in Fig. 




3. Experiment
Rhodamine101 (Rh101) was purchased from Sigma–Aldrich and the molecular skeleton is shown in the inset of Fig.
 | Fig. 3. (color online) (a) Normalized steady-state absorption (dashed curve) and fluorescence spectrum of Rh101+/MEOH, where inset shows molecular skeleton of Rh101; (b) typical TG spectrum of Rh101+/MEOH measured upon excitation at 532 nm. ESA band and GSA band as well as SE band can be observed in this spectrum. |
Steady-state absorption spectrum was measured with an UV–Vis spectrophotometer (720 PC, Shanghai). Steady-state fluorescence spectrum upon excitation by a 532-nm femtosecond laser was recorded with a spectrometer (Chromex 500IS/SM, BRUKER) and CCD (DU440, Andor).
The TG measurement step has been reported previously.[22] Briefly, the 800-nm, 110-fs pulse duration, 1-kHz repetition output pulse was split into two beams by a 9:1 beam splitter. The 90% beam pumped an optical parametric amplifier (OPA, OPA-800FC, Spectra Physics) to produce a pulse with a central wavelength of 532 nm, which was split into two uniform beams. The energy of each pulse is 0.3 νJ. These beams that are resonant with the sample were used as the pump light in the TG experiment. Because the bandwidth of femtosecond pump pulse was about 10∼nm, the coherent vibrational modes with low frequency could be stimulated coherently through impulsive stimulated Raman scattering.[23]
The other 10% beam was focused onto water to produce a WLC, which was used as the probe light. Its spectral range could cover the entire visible region. The probe light went through the optical delay line (ODL) and superposed with the two pump beams in the sample spatially. The TG signal appeared in the direction of the phase-matching condition and was transmitted to the spectrometer and measured by a thermoelectrically cooled charge coupled device (CCD) (DU440, Andor). The stepping motor in the ODL moved in steps of 62.5 fs to give time-resolved TG spectra. All of the experiments were carried out at room temperature in a dark room.
4. Results and discussion
To verify the dynamical information at different wavelengths in time-resolved BB-TG spectrum, the steady-state UV-Vis absorption spectrum and fluorescence spectrum of Rh101+/MEOH (mixed solution) are measured. The normalized steady-state absorption spectrum and the fluorescence spectrum (excited at 532 nm) of the sample are shown in Fig.
The excited state lifetime of Rh101+ in methanol is about 4 ns,[25,26] basically, the intensity of TG spectrum has slight change in our detecting time window (tens of ps). Typical TG spectrum is shown in Fig.
The high frequency vibrational modes of Rh101 have been discussed before by experimental measurement and calculation.[17,28] Moreover, due to limitation of the pump pulse duration of 110 fs, only components with a period longer than 220 fs (frequency less than 150 cm−1) can be distinguished efficiently according to the Nyquist sampling theory. So, we only focus on the low-frequency portion of the TG spectrum in the following analysis. For the solvent, the lowest frequency vibration mode of MEOH is larger than 400 cm−1, which will not obstruct the identification of low-frequency modes of Rh101+ in FT power spectra.
The TG transient in SE & GSA overlap band (585 nm) and ESA band (460 nm) is shown in Fig.
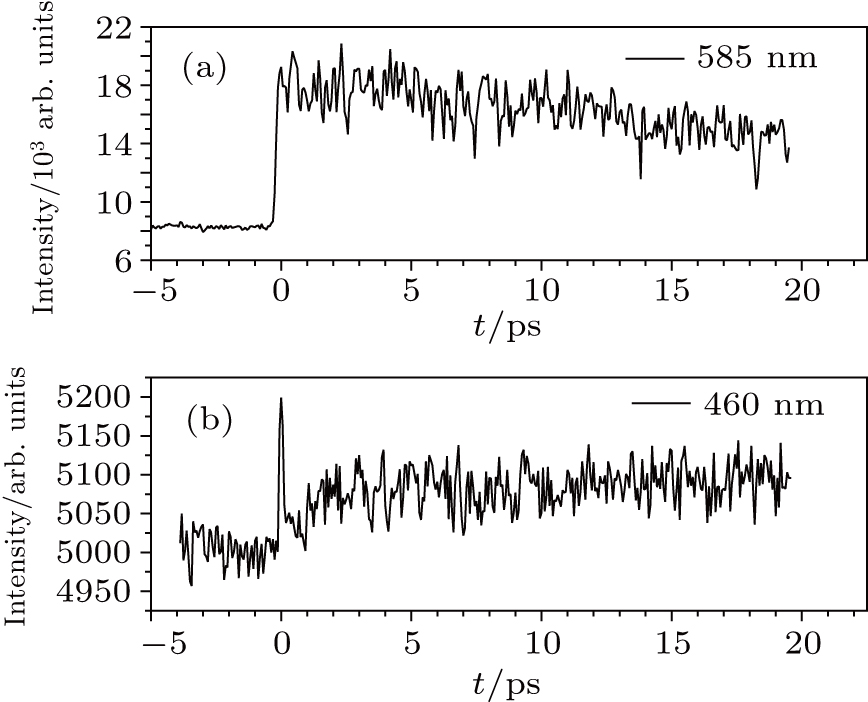 | Fig. 4. TG transient of Rh101+/MEOH in (a) GSA & SE overlap band and (b) ESA band. |
The FT power spectra of the TG signal at 460 nm and 585 nm in positive range are shown in Figs.
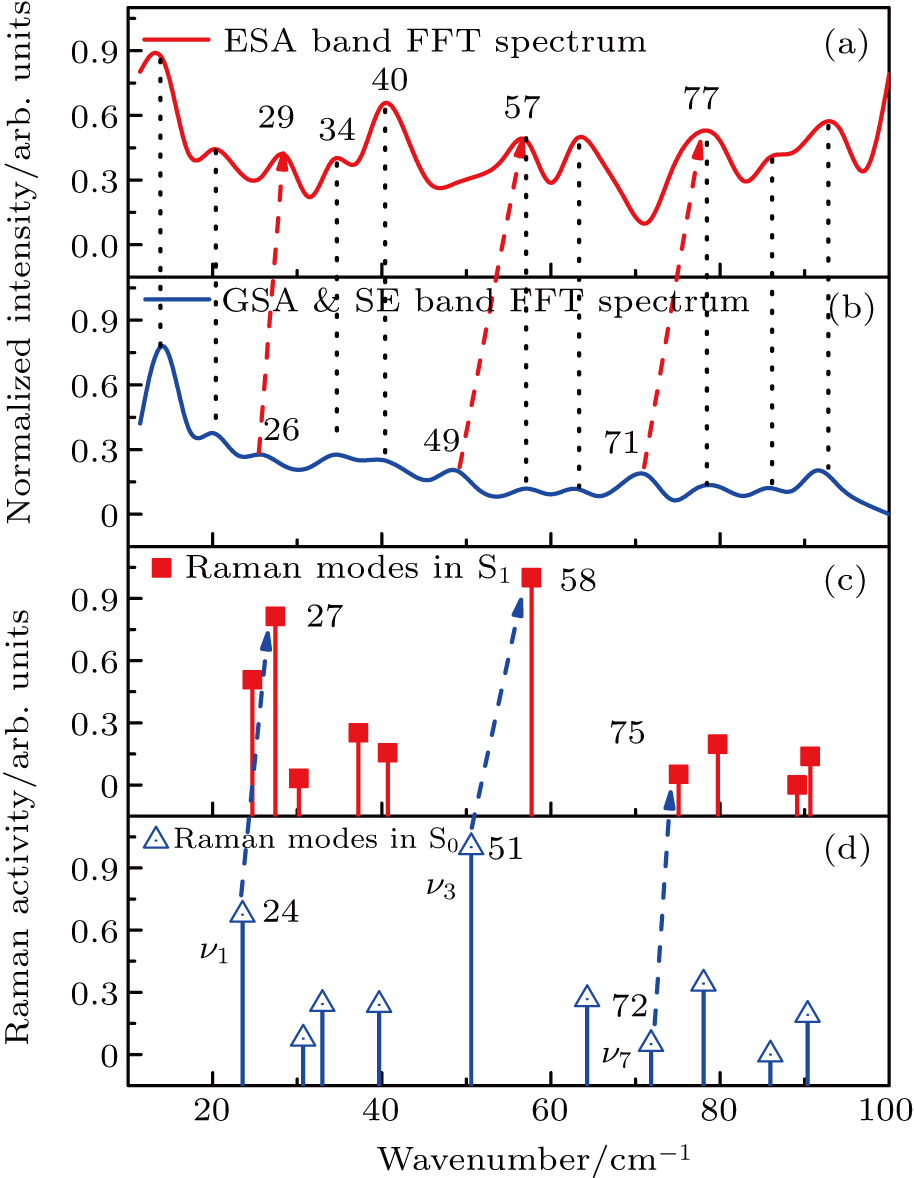
 | Fig. 5. (color online) (color online) Fourier transform power spectra in (a) ESA band-460 nm and (b) GSA & SE band-585 nm; Gaussian calculated Raman modes in minimum structure of (c) S1 state and (d) S0 state. Black-dotted lines connecting panels (a) and (b) represent the same vibrational modes. Dashed-blue arrows pointing to panel (c) from panel (d) denote calculated same vibrational mode blue shift from electronic ground state to excited state while the dashed red arrows refer to the corresponding experimental results. |
To identify the vibrational coherence in different electronic states, the quantum chemical calculation program Gaussian 03[29] is adopted. The minimum energy point configuration of ground and first electronic excited states of Rh101+ radical cation are optimized by the density functional theory (DFT) and TD-DFT B3LYP method with the “6-311+G (d, p)” basis set. The solvent effect is treated by the polarizable continuum model (PCM), methanol is chosen as the solvent to keep consistent with the experiment. The calculated Raman frequencies of Rh101+ in ground state and excited state are not scaled in this work because the Raman scaled factor was not suited for all of the vibrational modes.[30,31] Moreover, a few percent correction has little influence on low-frequency modes that are under 100 cm−1.
The calculated vibrational modes of Rh101+ in S1 and S0 states are shown in Figs.
| Table 1. Vibrational modes of Rh101+ in S0 and S1 states and their assignments based on calculations. . |
The experimental results are consistent with theoretical expectations. In Figs.
Three vibrational modes’ blue shifts are apparent in the FT spectra, as denoted by dashed-arrowed upwards lines in Fig.
Other distinct modes’ blue shifts also appear in the FT power spectra. First, the wag mode of the phenyl ring parallel to xanthene ring (ν5) with a frequency of 49 cm−1 in the S0 state only arises in the GSA & SE band FT power spectrum, while the same mode in S1 state with a frequency of 57 cm−1 appears both in GSA & SE and ESA FT power spectra. This result coincides with the calculated change from 51 cm−1 to 58 cm−1 within the error range. Second, the calculated C–C–C ring anti-symmetrical twist vibration (ν7) with a frequency of 72 cm−1 in S0 state only appears in the GSA & SE band FT spectrum as indicated at 71 cm−1, while the calculated same mode in S1 state with a frequency of 75 cm−1 coupled with 80 cm−1 (78 cm−1 in S0 state) vibration mode occurs in both of two FT spectra with frequencies of 77 cm−1 and 80 cm−1.
The peaks at both 14 cm−1 and 20 cm−1 only appear on the two FT power spectra; however, the calculated results show that there is no molecular vibrational mode with frequency below 23 cm−1, which demonstrates that none of them is derived from single molecular vibration but may originate from collective vibrations. The FT power spectrum at about 30 cm−1 appears in neither of the two states. and the FT power spectrum near 25 cm−1 in S0 state may come from their relatively low Raman activity. In addition, the unexpected envelop occurs at 63 cm−1 in Fig.
Through a comparative analysis between quantum chemical calculation and FT spectra of TG signal, it can be confirmed that the vibrational information in ground- and excited-state of Rh101+ can be separated by BB-TG spectra and their FT power spectra at various wavelengths. Specifically, the FT spectrum in GSA & SE band contains the mixed dynamics of ground and excited state while excited state vibrational information is included only in ESA band.
Furthermore, according to the quantum chemical calculation, it is also found that the molecular configuration of Rh101+ is transformed during the electronic transition from S0 to S1 state. In brief, the xanthene ring, phenyl and the carboxyl group show better coplanarity after electronic transition. Interestingly, the vibrational modes of Rh101+ involving with phenyl and carboxyl group present relatively obvious changes during the electronic transition from S0 to S1 state; for instance, ν1, ν3, ν5 and especially ν6 shift 4, 8, 7, and 40 cm−1, respectively. The other modes shift inconspicuously; for example, ν2, ν10 shift less than 1 cm−1, even the most remarkable mode (ν7) only shift less than 3 cm−1 (the carboxyl group torsion in ν8 is quite weak). The geometry change of Rh101+ in S1 state concentrates on phenyl and carboxyl group, and the results reveal that molecular configuration has a decisive influence on the frequency of the same vibrations.
5. Conclusions
Theoretical analysis based on double-sided Feynman diagrams indicates that the dynamics in GSA & SE overlapped band contains both ground and excited electronic states’ vibrational information. Meanwhile, only excited electronic state vibrational coherence is contained in the ESA band. The TG experiment in which a broadband probe pulse with Rh101+ is used as a target molecule is carried out. GSA & SE overlapping band and ESA band are both observed in the time-resolved TG spectra. After Fourier analysis at particular wavelengths in positive delay time range of the TG dynamics and comparing with quantum chemical calculation, the theoretical expectations are proven. Low-frequency vibrational modes of Rh101+ in ground- and excited- electronic states are distinguished. Three vibrational modes with obvious blue shift are observed after the electronic transition. The results also reveal that the frequency of specific vibrational mode is seriously affected by the molecular structure and by the electronic states.
This work proves that the Fourier transform (FT) for TG transient at various wavelengths is an effective way to distinguish vibrational modes from different electronic states. This method shows good performance, even when the frequency of vibration mode is relatively low and the target molecule has strong fluorescence. Owing to its simple operation and relative high efficiency, this method can be generalized to more molecules.
Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] |