She Yan-Chao, Wei Zhao, Luo Kai-Wu, Li Yong, Zhang Yun, Zhang Wei-Xi. Electronic and magnetic properties of semihydrogenated, fully hydrogenated monolayer and bilayer MoN2 sheets*
Project supported by the National Natural Science Foundation of China (Grant Nos. 11747168, 11604246, and 11704007), the Natural Science Foundation of Guizhou Provincial Education Department, China (Grant Nos. KY[2015]384, KY[2015]446, and KY[2017]053), the Natural Science Foundation of Guizhou Provincial Science and Technology Agency (Grant Nos. LH[2015]7232 and LH[2015]7228), and the Science Research Foundation of Tongren University, China (Grant No. trxyDH1529).
. Chinese Physics B, 2018, 27(6): 060306
Permissions
Electronic and magnetic properties of semihydrogenated, fully hydrogenated monolayer and bilayer MoN2 sheets*
Project supported by the National Natural Science Foundation of China (Grant Nos. 11747168, 11604246, and 11704007), the Natural Science Foundation of Guizhou Provincial Education Department, China (Grant Nos. KY[2015]384, KY[2015]446, and KY[2017]053), the Natural Science Foundation of Guizhou Provincial Science and Technology Agency (Grant Nos. LH[2015]7232 and LH[2015]7228), and the Science Research Foundation of Tongren University, China (Grant No. trxyDH1529).
She Yan-Chao1, Wei Zhao2, Luo Kai-Wu1, Li Yong1, Zhang Yun2, †, Zhang Wei-Xi1, ‡
Department of Physics and Electronic Engineering, Tongren University, Tongren 554300, China
Department of Physics and Information Technology, Baoji University of Arts and Sciences, Baoji 721016, China
Project supported by the National Natural Science Foundation of China (Grant Nos. 11747168, 11604246, and 11704007), the Natural Science Foundation of Guizhou Provincial Education Department, China (Grant Nos. KY[2015]384, KY[2015]446, and KY[2017]053), the Natural Science Foundation of Guizhou Provincial Science and Technology Agency (Grant Nos. LH[2015]7232 and LH[2015]7228), and the Science Research Foundation of Tongren University, China (Grant No. trxyDH1529).
Abstract
Based on density functional theory, we investigate the electronic and magnetic properties of semi-hydrogenated, fully hydrogenated monolayer and bilayer MoN2. We find that the AB stacking bilayer MoN2 exhibits ferromagnetic coupling of intralayer and antiferromagnetic coupling of interlayer, however, the ground states of the semi-hydrogenated, fully hydrogenated monolayer and AA stcaking bilayer MoN2 are nonmagnetic. The fully hydrogenated system has a quasi-direct band-gap of 2.5 eV, which has potential applications in light-emitting diode and photovoltaics. The AB stacking bilayer MoN2 shows the Dirac cone at K point in BZ around Fermi energy. Furthermore, the interlayer of the AB stacking bilayer MoN2 is subjected to a weak van der Waals force, while the interlayer of the AA stacking forms N-N covalent bond.
Very recently, the nanoscience and emerging nanotechnologies have been dominated by the honeycomb carbon structures in different dimensionalities, such as fullerenes, single (multiwalled) carbon nanotubes, graphene and its ribbons. In particular, graphene with two-dimensional (2D) honeycomb structure is one of the most intriguing carbon structures, in which sp2 hybridized electrons (σ electrons) form a honeycomb structure and the remaining pz (π) electrons follow the massless Dirac (Weyl) equation.[1–4] The energy bands show the linear dispersion (Dirac cone) at the Fermi level (EF) at particular symmetry points, K and K′, in the Brillouin zone (BZ).[5] This gives rises to unique phenomena such as the anomalous quantum Hall effect and unexpected magnetic ordering,[6] which make graphene a promising material used for the next-generation nanodevices.[7] However, graphene with flat surfaces is nonmagnetic, which has limited their potential applications in spintronics. Until now, obtaining the ordered spin structures in 2D materials that exist in ambient conditions is a great challenge.
Recently, transition-metal dinitrides (TMDNs) with rhombohedral MoS2 structure have been experimentally synthesized,[8] where Mo layer is sandwiched between two sulfur layers by covalent forces. Radisavlijevic et al. have demonstrated that it exhibits high on-off current ratio and good electrical performance, which may be used in the future electronic circuits requiring low stand-by power.[9] The strong emission inherited from the direct gap structure of monolayer MoS2 also promises the applications in optoelectronics.
The chemical functionalization is an effective way to tailor the electronic structures of nanostructures. Hydrogen is lowly electronegative and can be used as a functionalization group in nanostructures, such as graphene, germanene, silicon carbide, and boron nitride.[10–13] Recently, the studies of the two-dimensional fully functionalized and semifunctionalized sheets have aroused lots of interest of all.[14–16] However, the influence of the hydrogen-functionalization on the MoN2 sheet has not been mentioned yet. Thus, we focus our attention on the MoN2 sheet functionalized with hydrogen. In this work, we present a systematic investigation on the electronic and magnetic properties of semihydrogenated (MoN2–H), fully hydrogenated (H–MoN2–H) monolayer and bilayer MoN2 sheets.
2. Calculation methods
Density functional theory (DFT) calculations were carried out by using the projector augmented wave method and the generalized gradient approximation (GGA), as implemented in the Vienna ab initio simulation package (VASP).[17–19] The exchange–correlation functional, vdW-DF, which is capable of treating the van der Waals (vdW) force,was adopted (optB86-vdW functional).[20–22] The projector augmented wave pseudopotential with an energy cutoff of 400 eV for the plane-wave basis sets was adopted, and Gamma-centered 15 × 15 × 1 Monkhorst–Pack k-point meshes were used for slab calculations.[23,24] The convergence criteria were set to be 1 × 10−5 eV for total energy and 0.005 eV·Å−1 for Hellman-Feynman force, respectively. A vacuum region of 25 Å was applied in the z direction, which is large enough to make the interaction between neighboring images negligible. Ab initio molecular dynamics simulations with canonical ensemble (NVT) at the temperatures of 300 K and 600 K are performed in 3 ps, respectively. A supercell containing 4 × 4 unitcells is adopted as the model.
3. Results and discussion
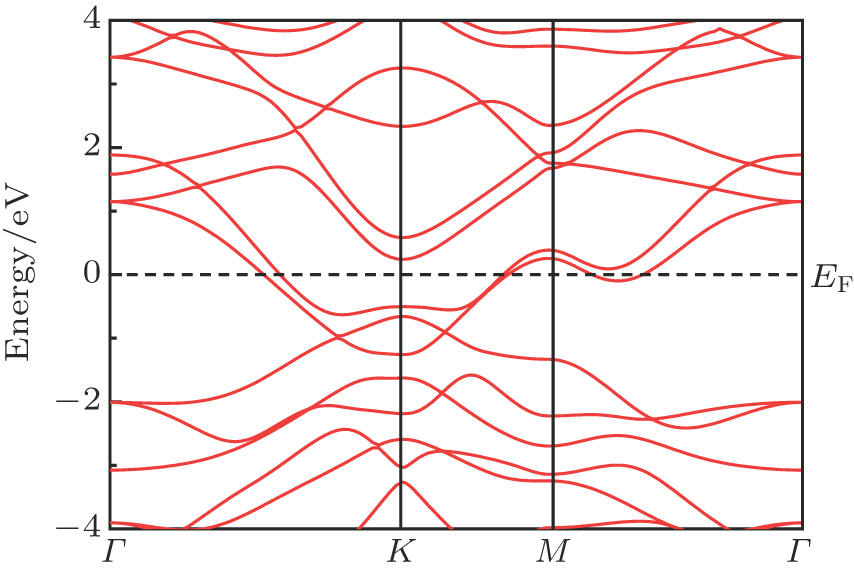
The bulk 3R-MoN2 belongs to a rhombohedral R3m space group and has a layered structure. The interaction between the MoN2 layers is weak van der Waals force, with small exfoliation energy; hence it has been proposed that the monolayer MoN2 can be exfoliated using similar methods to those for other 2D materials. As shown in Fig. 1(a), the monolayer MoN2 is of hexagonal lattice symmetry, and each nitrogen ion is surrounded by three Mo ions, forming a pyramid-shaped structure. In order to verify the accuracy of our calculations, we firstly calculate the lattice parameter of the pristine MoN2 sheet. The calculated lattice parameter is a = 2.98 Å, and the Mo–N bond length is 2.05 Å, which is in good agreement well with previous theoretical values, but smaller than that for the Mo–S distance (∼ 2.41 Å) in MoS2 due to the smaller ionic radius of N.[25,26] We note that the N–N bond between the two N sites is along the vertical direction. The bond length dN−N is 2.23 Å, which is longer than the typical N–N bond length (∼ 1.45 Å), resulting in a negligible bonding interaction. As the electronic band structure is shown in Fig. 1(b), the MoN2 sheet presents a metallic property, which also accords with previous calculations.[25,26] The energy of the ferromagnetic (FM) configuration is about 0.24 eV per formula unit lower than that of the spin unpolarized configuration. The magnetic moment is mainly from the N (2p) sublattice (∼ 0.42 μB), whereas the Mo (4d) moment is much smaller (∼ 0.13 μB). Consideing the three empty p orbitals in each nitrogen ion, we obtain the picture that Mo ions cannot effectively saturate the 2p orbital of N ions, thus leading to the spontaneous magnetic moments at N sites. Most of local magnetic moments are contributed by the N pz orbitals.
Fig. 1. (color online) (a) Monolayer MoN2 (2 × 2 supercell). (b) Calculated energy band of the stable structure for FM state MoN2. Black and red color indicate the spin-up and spin-down. Mo and N atoms are in blue and red, respectively.
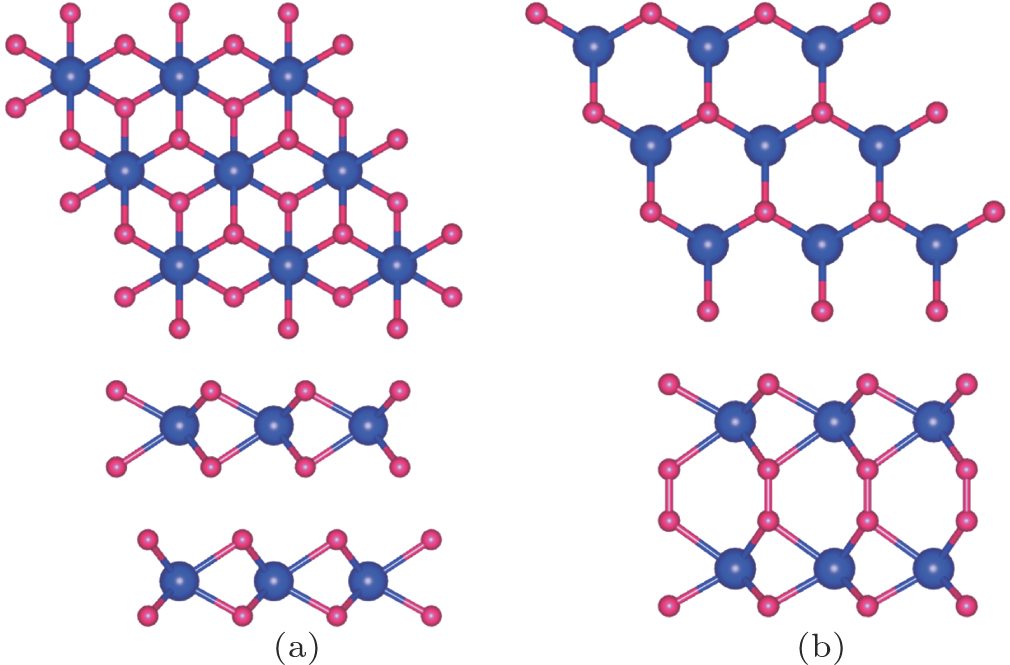
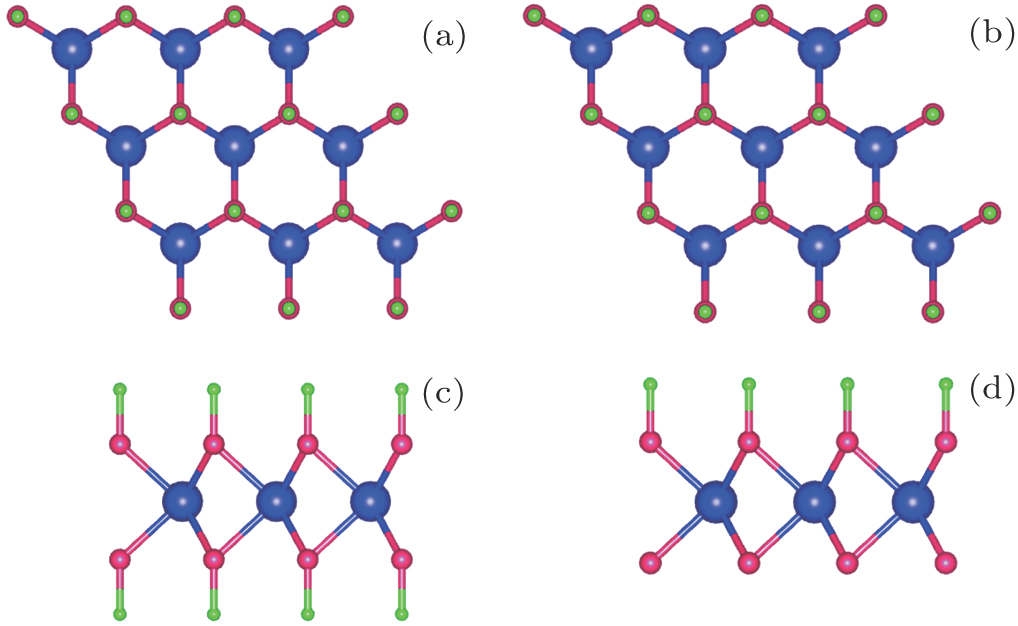
We firstly discuss the results of hydrogenated MoN2 sheet (H–MoN2–H), which is similar to its carbon counterpart, graphene, as shown in Fig. 2(a). The distance between Mo and N is found to be 2.10 Å. The hydrogen atoms are absorbed on the top site of N atoms and H-N bond lengths each are 1.02 Å. To quantify the stabilization of H–MoN2–H, we calculate the formation energy Ef = (Etot − EMoN2 − (NH/2)EH2)/NH. Here Etot, and EMoN2 are the total energies of the H–MoN2–H and the MoN2, respectively; EH2 is the energy of H2 which is chosen to be the total energy of an isolated H2; NH is the number of H atoms in the H–MoN2–H. The formation energy is −1.808 eV/H, showing that H–MoN2–H is stable.
Fig. 2. (color online) (a) Fully hydrogenated monolayer MoN2 (2 × 2 supercell). (b) Semi-hydrogenated monolayer MoN2 (2 × 2 supercell). Mo, N, and H atoms are marked in blue, red and green, respectively.
In H–MoN2–H configuration, we find that the system has a nomagnetic moment for each atom. The band structure is plotted in Fig. 3(c), and it is found that H–MoN2–H is a semiconductor with a direct band gap of 1.84 eV, which is different from the pristine MoN2 sheet. Both the valence band maximum (VBM) and conduction band minimum (CBM) are located at the K point in the Brillouin zone. There are some similarities between fully hydrogenated graphene and MoN2 sheet. Hydrogenation opens their band gaps. Further, the calculated optical spectra of H–MoN2–H with and without excitonic effects are shown in Fig. 4. The exciton effect (red solid line) dielectric function is red-shifted. We find that the optical absorption starts at 2.5 eV. It is shown that H–MoN2–H is a very good solar cell absorber and has potential applications in light-emitting diode and photovoltaics.
Fig. 3. (color online) Calculated results of MoN2–H and H–MoN2–H. (a) Three magnetic coupling states. ((b) and (c)) Calculated energy bands of the stable structures for the MoN2–H and H–MoN2–H. EF is shown by dashed lines.
Fig. 4. (color online) Calculated imaginary part of the dielectric function of H–MoN2–H with (red solid line) and without (black solid line) excitonic effects.
As stated in previous discussion, the hydrogenation of MoN2 sheet is an exothermic process. Once fully hydrogenated, H can bind on N site with a bond length of 1.02 Å for the corresponding N–H dimer in free state. Now the next question is starting from the fully hydrogenated MoN2 sheet. Can one find a way to remove hydrogen from one side forming a semihydrogenated MoN2 sheet? If so, how will the properties change?
Like the graphene sheet, all the N sites are equivalent. Thus, when removing half of H atoms from a fully hydrogenated MoN2 sheet, we have only one option for the semihydrogenated sheet as shown in Fig. 2(b). The optimized structure shows that the Mo–N bond length is reduced to 2.02 Å on the site without hydrogen atoms. While the Mo-N bond length is elongated to 2.13 Å in which the N atoms are hydrogenated, which is still comparable to the bond length of 2.10 Å of fully hydrogenated MoN2 sheet. These results are similar to those found in graphene. In MoN2–H configuration, we find that the magnetic moments are mainly from N atoms. To verify the ground state of this system, we calculate the total energies for the three magnetic coupling configurations on N atoms [Fig. 3(a)]: (i) FM coupling, (ii) antiferromagnetic (AFM) coupling, and (iii) nonmagnetic (NM) coupling. The results show that the NM coupling is 0.16 eV and 0.18 eV lower in energy than AFM and FM configuration coupling, respectively. The energy band structure is plotted in Fig. 3(b). We find that the system is metallic.
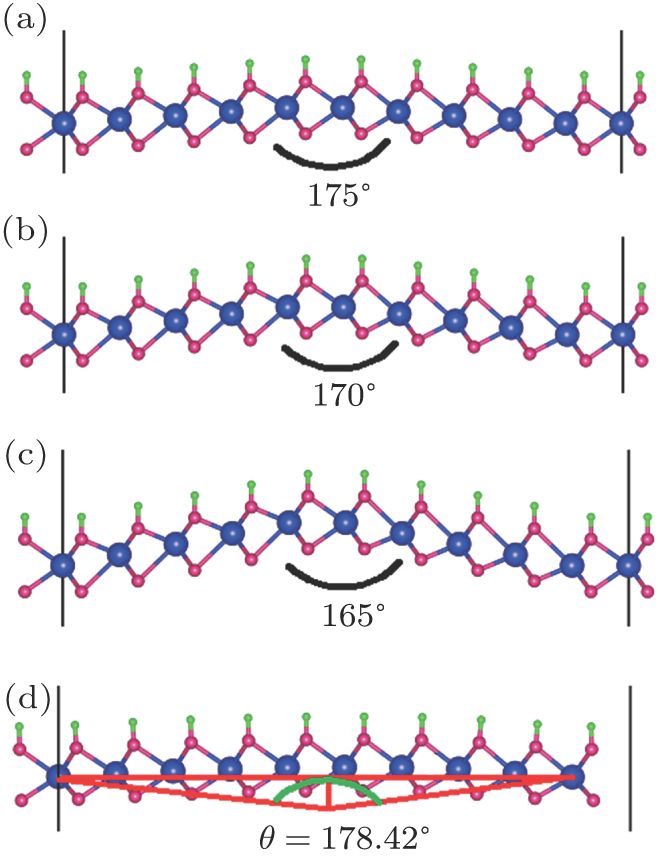
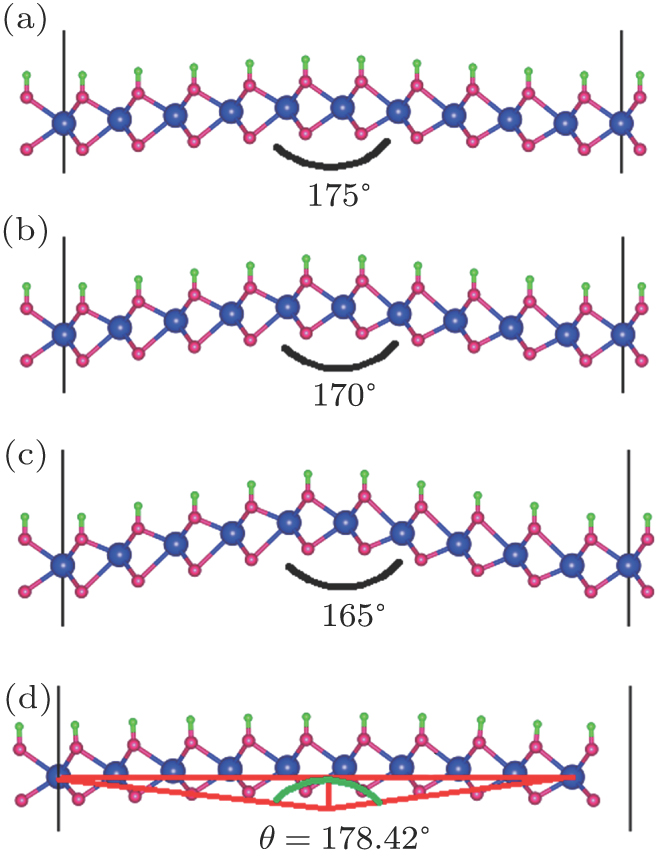
The semihydrogenated monolayers are asymmetric in the normal direction, therefore, it should be subjected to spontaneous bending. In order to estimate its intrinsic curvature, we calculate three bending structures as shown in Figs. 5(a)–5(c). After full relaxation, we find that these final structures are entirely the same as those shown in Fig. 5(d). The total energy of this structure is 0.43 eV lower than that of the semihydrogenated flat structure. We calculate the intrinsic curvature radius R = l/sin(θ/2) ≈ 13.39 Å.
Fig. 5. (color online) Three initial blending structures ((a)–(c)) before and (d) after relaxation.
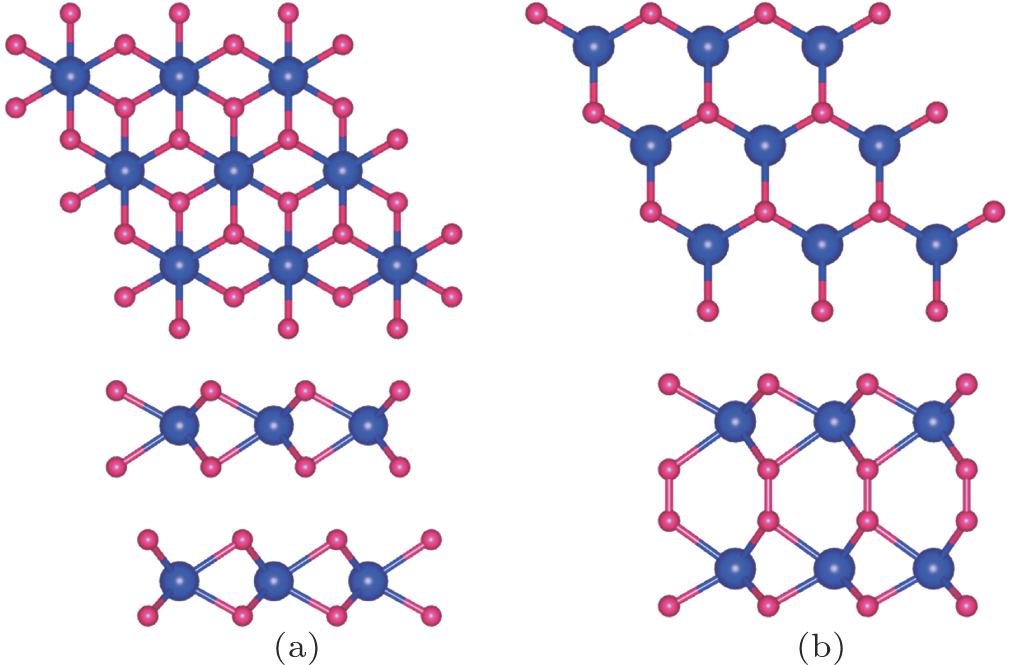
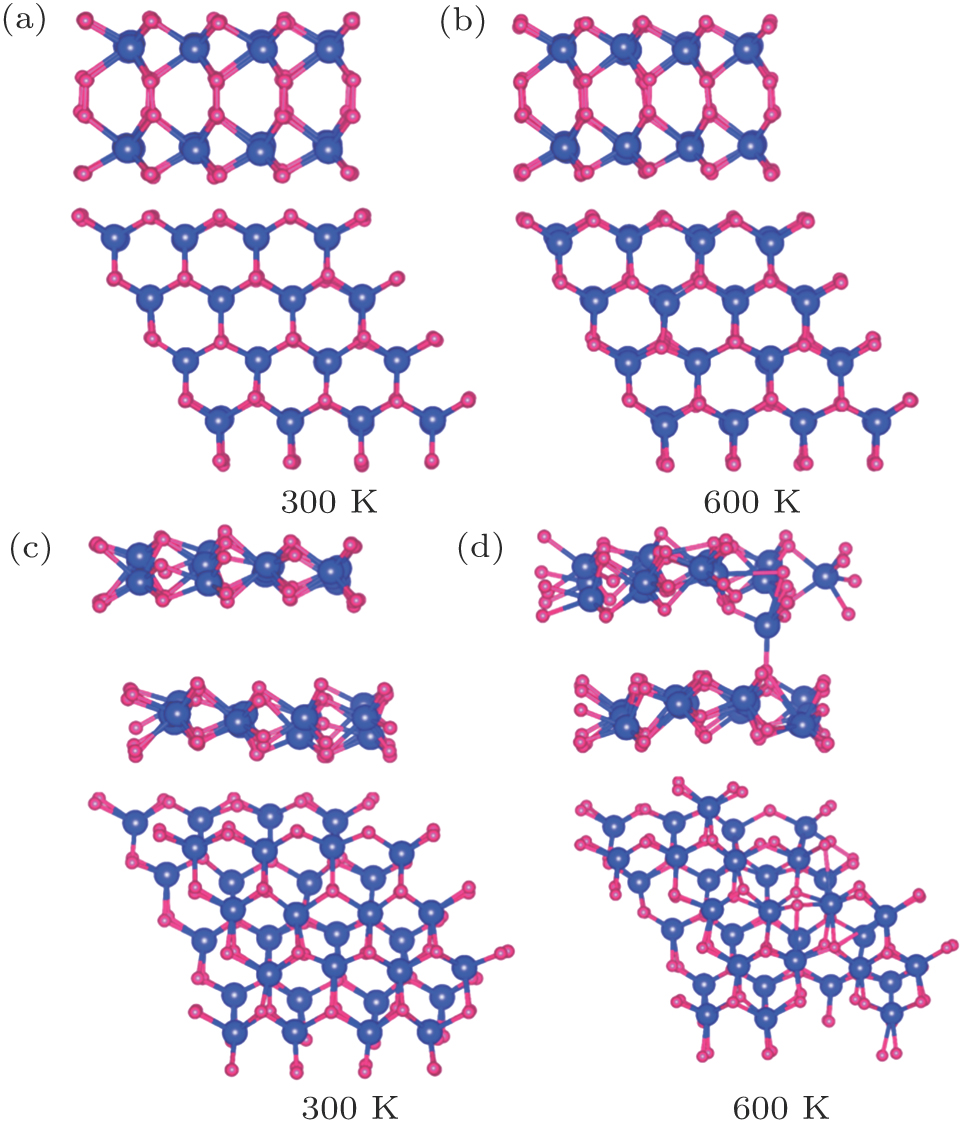
We now turn to the magnetic and electronic structure of bilayer MoN2 sheet [Fig. 6]. To verify that AA and AB stacking structure will be stable in ambient condition, ab initio molecular dynamics simulations at the temperatures of 300 K and 600 K are performed in 3 ps, respectively. Structure snapshots taken at the end of each simulation are shown in Fig. 7. Clearly, the planar structure of AA stacking is generally kept up to 600 K, indicating that AA stacking structure has good stability above the room temperature. The results show that the AB stacking MoN2 can be maintained at room temperature. We explore the magnetic origin of AB stacking MoN2. The intralayer spin coupling is ferromagnetic, whereas interlayer spin coupling is antiferromagnetic. The spin density of state shows that spin moments mainly come from the pz orbits of nitrogen ions. The magnetic moments of N and Mo atom are 0.13 μB and 0.27 μB, respectively. Interlayer coupling is ferromagnetic because the next nearest neighbor magnetic moments can interact with each other through bonding with the adjacent atoms. On the other hand, because of the overlapping pz orbital intralayer coupling is antiferromagnetic.
Fig. 6. (color online) (a) AB stacking bilayer MoN2 (2 × 2 supercell) and (b) AA stacking bilayer MoN2 (2 × 2 supercell). Mo and N atoms are in blue and red, respectively.
Fig. 7. (color online) Structure snapshots of ((a) and (b)) AA and ((c) and (d)) AB stacking MoN2 for MD simulation at 300 K and 600 K.
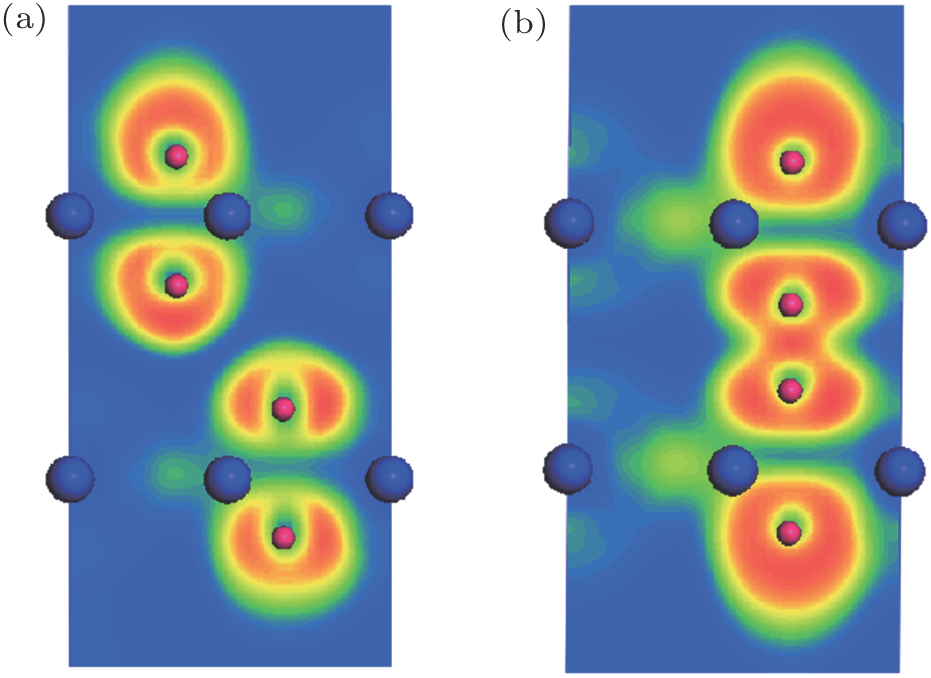
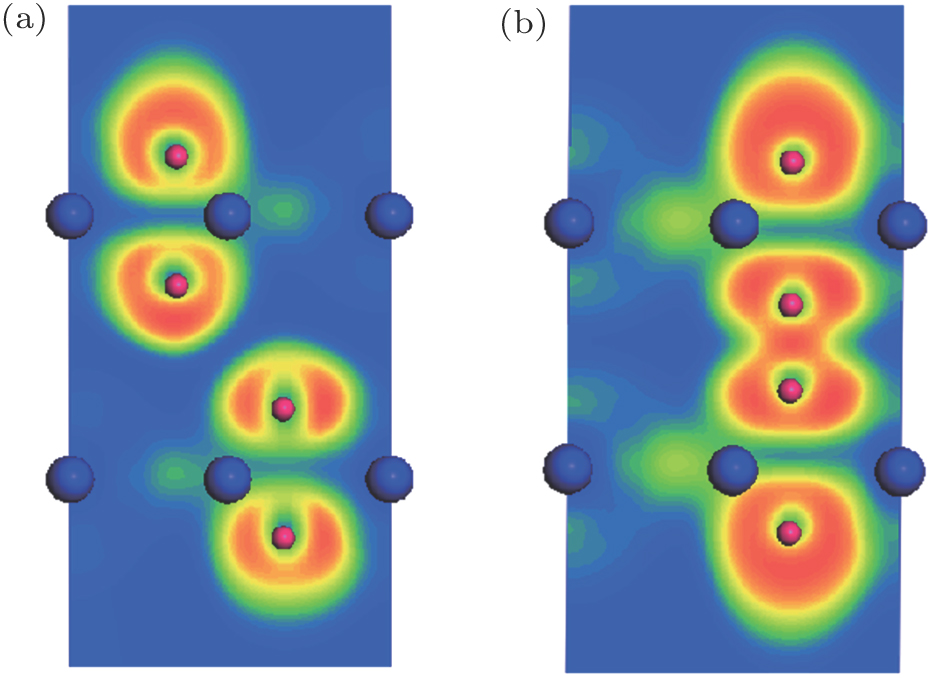
In the AA stacking bilayer MoN2 system, the interlayer coupling is nonmagnetic as a result of the N atoms bonding. In the AA stacking, dN−N = 1.38 Å, which is less than the typical N–N bond length (∼ 1.45 Å), implying the N–N single bond state. To verify the above schematic, we calculate the electron localization function (ELF) of the stable structures for AB stacking MoN2 and AA stacking MoN2, and the results are shown in Fig. 8. The ELF indicates the degree of electron localization and is useful in the analysis of bonding characteristics. We plot the ELF map in the vertical plane containing the four N atoms and two Mo atoms. In the AB stacking, it is clear that there is no bonding between the two N ions of intralayer. In contrast, a covalent bonding character appears between the two N sites of intralayer in AA stacking, indicating the formation of an N-N bond. We calculate the interlayer binding energy Eb = 2EMoN2 − Etot. Here EMoN2 and Etot are the total energies of the MoN2 and the AB stacking MoN2, respectively. The interlayer binding energy is 0.238 eV, indicating that it is due to the van der Waals interaction.
Fig. 8. (color online) Calculated electron localization functions (ELFs) of stable structures for (a) AB stacking MoN2 and (b) AA stacking MoN2. Plot is of the vertical plane cutting through two Mo atoms and four N atoms. Results indicate N–N nonbonding for AB stacking and bonding for AA stacking. Mo and N atoms are in blue and red, respectively.
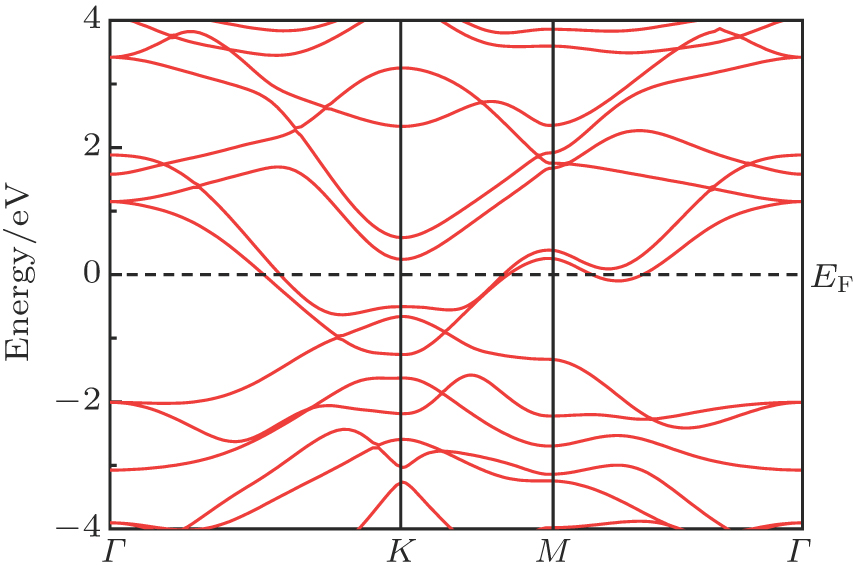
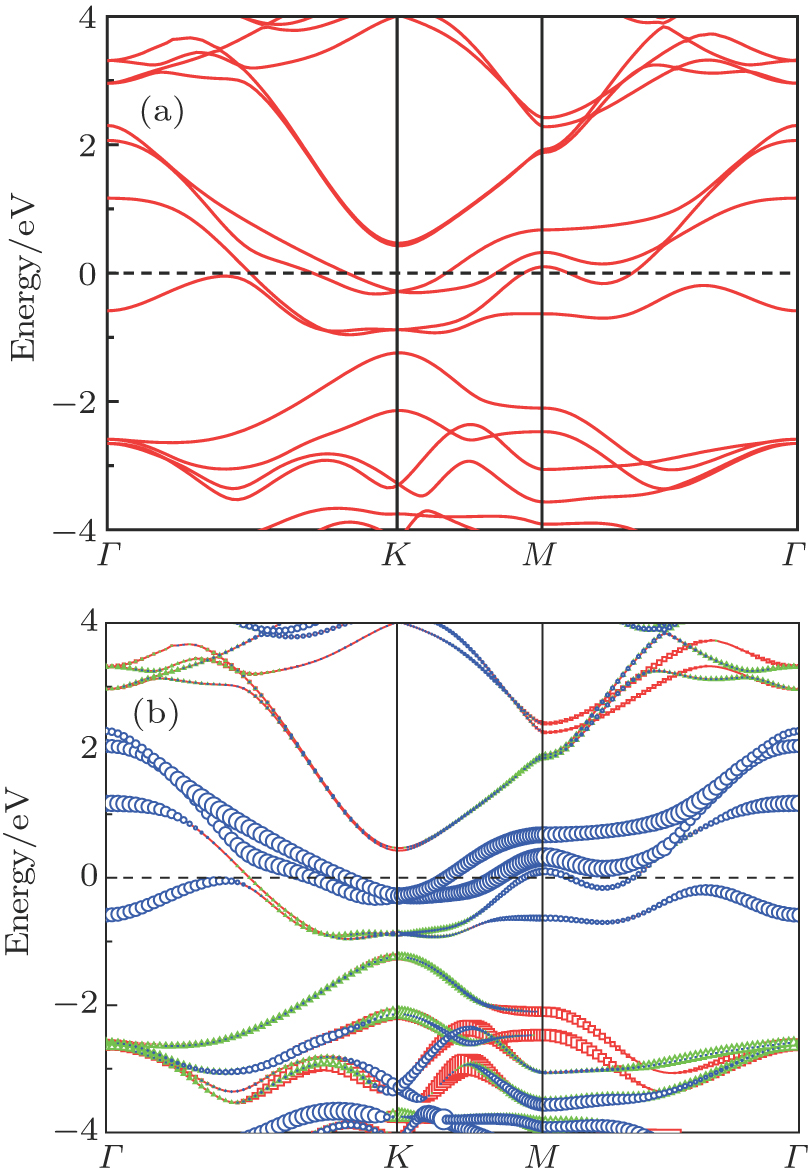
To find how stacking affects the electronic structure, we calculate the band dispersion for AB stacking and AA stacking as shown in Figs. 9 and 10. Although AFM materials are, in general, insulators, AB stacking is metallic. Moreover, AA stacking is also metallic. Interestingly, we find that in the AB stacking MoN2 there exists a linear energy band at K point in BZ around EF. Through the analysis of orbital projections on energy bands, it is found that the linear bands have almost entirely N 2pz orbital character (blue circles in Fig. 10(b)). The contribution of other states of N is extremely weak. This clearly shows that it is Dirac cone.
Fig. 10. (color online) Calculated energy bands for stable structure of AB stacking MoN2 in the cases (a) without orbital projections and (b) with orbital projections. Orbital projections for 2px (red square), 2py (green triangle), and 2pz (blue circle) on energy bands of MoN2 are also indicated.
4. Conclusions
In this work, we have systematically investigated the electronic and magnetic properties of semihydrogenated, fully hydrogenated monolayer MoN2 and bilayer MoN2 sheet. The main conclusions are as follows. (i) Our results reveal that the AB stacking bilayer MoN2 exhibits ferromagnetic coupling of intralayer and antiferromagnetic coupling of interlayer, however, the ground states of the semi-hydrogenated, fully hydrogenated monolayer and AA stcaking bilayer MoN2 are nonmagneticthe. (ii) The fully hydrogenated system has quasi-direct band-gap of 2.5 eV, showing that it is very good nanoscale solar cell absorbers. (iii) In bilayer MoN2, the interlayer of the AB stacking bilayer MoN2 is due to weak van der Waals force, while the interlayer of the AA stacking forms N–N covalent bond.
Electronic and magnetic properties of semihydrogenated, fully hydrogenated monolayer and bilayer MoN2 sheets*
Project supported by the National Natural Science Foundation of China (Grant Nos. 11747168, 11604246, and 11704007), the Natural Science Foundation of Guizhou Provincial Education Department, China (Grant Nos. KY[2015]384, KY[2015]446, and KY[2017]053), the Natural Science Foundation of Guizhou Provincial Science and Technology Agency (Grant Nos. LH[2015]7232 and LH[2015]7228), and the Science Research Foundation of Tongren University, China (Grant No. trxyDH1529).
[She Yan-Chao1, Wei Zhao2, Luo Kai-Wu1, Li Yong1, Zhang Yun2, †, Zhang Wei-Xi1, ‡]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}