


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Study of magnetic and optical properties of Zn1−x
TMx
Te (TM = Mn, Fe, Co, Ni) diluted magnetic semiconductors: First principle approach
Cite this Article
Mahmood Q, Hassan M, Faridi M A. Study of magnetic and optical properties of Zn1−x
TMx
Te (TM = Mn, Fe, Co, Ni) diluted magnetic semiconductors: First principle approach. Chinese Physics B, 2017, 26(2): 027503 
Permissions
Study of magnetic and optical properties of Zn1−x
TMx
Te (TM = Mn, Fe, Co, Ni) diluted magnetic semiconductors: First principle approach
† Corresponding author. E-mail:
Abstract
We present structural, magnetic and optical characteristics of 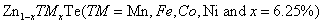
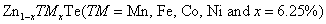
Keyword:magnetic semiconductors;density functional theory;optical and dielectric properties;electron density of states and band structure of crystalline solids
1. Introduction
A wide range of theoretical and experimental reports, available in the literature, depict huge motivations to explore interesting materials for versatile device functionalities.[1–10] The semiconductors belonging to II–VI, III–VI, IV–VI, are extensively being explored for various applications. Particularly, the II–VI based DMSs, for example ZnS, ZnSe, and ZnTe, are considered to be attractive owing to their wide direct band gaps, which can offer versatile opto-electronic characteristics, therefore, could be expected to replace the existing silicon-based devices.[11–15] ZnTe crystallizes in zinc blende phase with a cubic lattice constant of 6.103 Å,[16] and the reported direct band gap is 2.26 eV,[17] which enables its application in the devices operable at high temperatures and power levels. ZnTe has attracted huge attention because of its low cost, large absorption coefficient, and higher solubility to magnetic impurities without significant structural deteriorations.[18,19]
The undoped and doped ZnTe in thin film, nanoparticle, and bulk forms have been fabricated and characterized with various experimental techniques.[20–25] For example, thin films of Zn1−x
Ni
x
Te (x
Ni up to 20%), deposited on glass substrate by employing electron beam evaporation, have demonstrated that with the increasing Ni content, the band gap and micro strains are reduced, while the grain size is improved, which depict that Ni impurity ions may induce optical and structural modifications.[26] The nanocrystals of Mn-doped ZnTe synthesized by hot injection method have shown that the emission band centered at 620 nm, corresponds to narrow absorption bands for different crystal sizes, which confirmed that emission is due to Mn+2 substituted ZnTe.[27] Similarly, the Co-doped ZnTe prepared by thermal diffusion method has depicted that Co doping shifts the absorption edge towards the lower energy, and optical transitions of the electrons to the dopant induced states are considered to be responsible for the observed absorption lines.[28] Another report about Fe-doped ZnTe (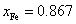
The ZnTe has widely been explored by using the density functional theory (DFT). In a previous report on ZnTe doped with 25% Fe, Ni, Co, samples have exhibited half-metallicity due to Fe and Ni doping, while semiconducting properties are attained due to Co doping.[30] The results are found to be consistent with a previous comprehensive theoretical study, in which Cr, Fe and Ni doping in ZnTe results in a half-metallic character, while Co and Mn depict semiconducting behavior.[31] The half-metallic ferromagnets (HMFMs), in which one sub-band show metallic nature, while the other sub-band exhibit insulating behavior, and the semiconducting materials, in which both sub-bands show insulating behaviors, are crucial for novel spintronic applications.[31,32] Moreover, Mn- and V-doped ZnTe have also been investigated by Wein2K package and it observed that for 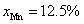

In this study, we report a comprehensive investigation of structural, electronic, magnetic and optical properties exhibited by Zn1−x
TMx
Te (

2. Method of calculations
Various novel material characteristics[39–41] can be elucidated by employing various theoretical methods.[42–44] In the present study, we have used FP-LAPW method, which is implemented in Wein2K software,[45] to compute the structural and electronic characteristics of Zn1−x
TMx
Te (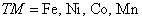

In order to control the plane-wave expansion, the convergence parameter represented as R
MT × K
max, (R
MT represents the smallest radius of the MT spheres, while K
max indicates the cutoff for the plane-wave), is taken as 7.0. The maximum values of the angular momentum, I
max, is taken as 10. To avoid the charge leakage, the separation between core and valence states is defined in terms of energy cut-off parameter, which is adjusted to −6.0 Ry (
3. Results and discussions
3.1. Structural stability
where E
Total (TMl
Zn
m
Te
n) represent the total ground state (GS) energy of TM-doped (
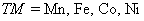
) ZnTe DMSs, while E
TM, E
Zn, and E
Te are the GS energies of bulk elements in a unit cell. The subscripts l, m, n, respectively, represent the number of TM, Zn, and Te ions in each unit cell volume. The formation energies exhibit negative sign (see Table 1 ) which indicates that energy releases during compound formation, elucidating stability in FM state. Moreover, the TM doping causes higher energy release, in contrast to the experimental value (−28.10 eV)[53] for ZnTe, as shown in Table 1 . Hence, TM dopants enhance the phase stability, which is an additional advantage. The stability of Zn1−x
TMxTe compounds is also confirmed from the cohesive energy (E
coh) that binds the atoms together in the crystal. The E
coh is computed by employing the following equation
The realization of a stable Zn-based diluted magnetic semiconductors (DMS) phase is a controversial topic being widely explored. Sato et al.[48] have shown that spin glass phase is present in Zn1−x
TMx
Te (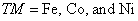
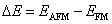
 | (1) |
 | (2) |
Enthalpy of formation ΔH
f (eV), ground state energy ΔE (meV), Curie temperature T
c (K), cohesive energy (E
coh), direct exchange splitting energy Δ
x
(d), indirect exchange splitting energy Δ
x
(pd), exchange constants (N
0
α and N
0
β, the total magnetic moment per unit cell, magnetic moment on TMs site, magnetic moment on Te-site, band gap (E
g), half metallic band gap (G
HM), static dielectric constant ε
1(0), and refractive index n(0) calculated for 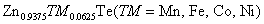
| Composition | Exp. | ZnTe | ZnMnTe | ZnFeTe | ZnCoTe | ZnNiTe |
|---|---|---|---|---|---|---|
| Δ H f/eV | −28.10 |
– | −94.08 | −95.22 | −94.19 | −94.64 |
| ΔE/meV | – | – | 4.10 | 3.72 | 4.41 | 3.10 |
| T c/K | – | – | 494 | 453 | 417 | 380 |
| E coh | 4.48 |
4.55 | 5.90 | 5.95 | 6.04 | 6.25 |
| (Δ E crystal | 2.07 | 1.33 | 2.92 | 0.86 | ||
| Δx(d) | 3.07 | 2.14 | 5.33 | 3.02 | ||
| Δx(pd) | −0.80 | −0.79 | −0.57 | −0.53 | ||
| N 0 α | −0.217 | 0.236 | −0.256 | 0.483 | ||
| N 0 β | −0.874 | −0.925 | −0.943 | −1.086 | ||
| Total MM | 5.00 | 4.00 | 3.00 | 2.00 | ||
| TM-site | 4.245 | 2.572 | 2.423 | 1.313 | ||
| Zn-site | 0.254 | 0.345 | 0.311 | 0.232 | ||
| Te-site | 0.122 | 0.134 | 0.121 | 0.251 | ||
| E g | 2.39 |
2.39 | 2.32 | 2.37 | 2.36 | 2.32 |
| G HM | – | – | – | 0.05 | – | 0.14 |
| ε1(0) | 7.30 |
6.70 | 6.77 | 7.14 | 9.00 | 28.6 |
| n(0) | 2.57 |
2.58 | 2.62 | 2.67 | 3.00 | 5.41 |
Ref. [53],
Ref. [54],
Ref. [55]
Here E
Total(TMa
Zn
b
Te
c
) is the total ground state energy of the compound and 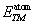
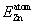
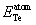
Moreover, Heisenberg model has been employed to estimate the Curie temperature of Zn1−x
TMx
Te compounds by using the expression 

3.2. Electronic and magnetic behavior
Here x shows TM ion concentration, 〈S〉 is the average value of magnetic moment on TM ion site,
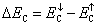
and

are the splitting at conduction and valence band edges, respectively. From Table 1 , it is clear that the N
0
α and N
0
β have anti-ferromagnetic coupling for Fe- and Ni-doped ZnTe compounds. While a ferromagnetic coupling for Mn- and Co-doped ZnTe compounds has been observed. The reason behind this anomaly can be explained in terms of valence and conduction band edge splittings. The kinetic exchange splitting at valence band maximum (VBM) results in large and negative N
0
β, while small and positive N
0
α appears at the conduction band minimum (CBM) because of the direct exchange splitting to generate half metallic ferromagnetic behavior. In FM semiconductors, the sign of N
0
α is strongly influenced by crystal symmetry and may have higher magnitude arising due to the quantum confinement. During the splitting process at CBM, it has been noted that d level lies below the CBM in the minority spin channel, due to which, the CBM in the minority and majority channels, respectively, will energetically be moved down and up and that results in negative N
0
α. On the other hand, half-metallic ferromagnetic semiconductors have been observed to have the d-levels lying below the CBM, for both the majority and minority channels. Hence, d-level is pushed up to the CBM toward high energy in both minority and majority spin,[68] which causes positive N0α, as shown by the values presented in Table 1 . Using the Heisenberg model, the Curie temperatures have also been predicted, which are 494, 453, 417, and 380 K for Mn-, Fe-, Co-, and Ni-doped ZnTe, respectively. The decrease in magnetic moment per magnetic ion from 5 μ
B to 2 μ
B by changing dopant from Mn to Ni causes a reduction in the magnitude of the exchange interactions and corresponding Tc reduces. Therefore, all the studied compounds can be used in various magnetic devices operating above room temperature.
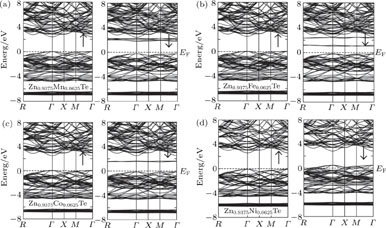
The modified Becke–Johnson potential (mBJ) along with generalized gradient approximation (GGA) are utilized to find the most accurate band structures (BS) as well as the precise density of states (DOS) in Zn1−x
TMx
Te (

 | Fig. 1. The band structure for both spin-up and spin-down channels plotted for (a) Zn0.9375Mn0.0625Te, (b) Zn0.9375Fe0.0625Te, (c) Zn0.9375Co0.0625Te, (d) Zn0.9375Ni0.0625Te, calculated by employing mBJ-GGA functional. |
 | Fig. 2. (color online) (a) The total (TDOS) and (b) partial density of states (PDOS) plotted for Zn0.9375TM0.0625Te (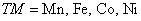
|
The real cause of the appearance of ferromagnetism has been investigated through the calculated partial density of states (PDOS), as presented in Fig. 
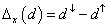
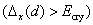
 | Fig. 3. (color online) The illustration of p–d repulsion model in Zn1−x Mn x Te. (I) Atomic magnetically polarized energy levels of Mn cation and host lattice Te anion, (II) exchange splitting of the energy levels Δ x (d), (III) further splitting of the Mn-3d energy levels due to the crystal field, (VI) p−d exchange splitting Δ x (p−d), due to the interaction between Mn-3d and Te-5p energy levels, the shaded area shows host lattice valence and conduction band. The figure description is followed by the reference.[59] |
In addition, during hybridization, the Mn and Co doping produce deficiency of electrons to induce hole-mediated ferromagnetism similar to that observed in GaMnAs, as is already explained by Zener.[61] In Fe- and Ni-doped ZnTe, the interaction arising between 3d-state of Fe/Ni impurities and p-states of host lattice Te anions, causes the appearance of the localized states within the band gap at E F, which induce half-metallic ferromagnetic characteristics. Therefore, the large splitting of t 2g states causes the double exchange mechanism that is responsible for producing ferromagnetism in Fe- and Ni-doped ZnTe half-metallic semiconductors.
Another parameter about exchange splitting energy is Δ
x
(pd), which is very important because its negative value shows that the p−d hybridization in the valence band is strong enough to realize a more attractive spin-down channel. The p−d repulsion model is used to justify the observed ferromagnetism, as is presented in Fig. 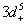
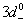

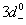
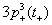
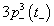
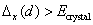




The number of unpaired electrons in 3d-state of TMs contributes to the observed magnetic moment. It is interesting to observe that Mn2+ (3d
5) has five unpaired electrons (two in e
g state and three in t
2g), Fe2+ (3d6) has four unpaired electrons (one in e
g state and three in t
2g), Co2+ (3d7) has three unpaired t
2g electrons and Ni2+ (3d8) has two unpaired t
2g electrons in their outermost shells, which are responsible for inducing free space magnetic moments.[64] Furthermore, the strong interaction between p-states of Te and dopant d-states reduces the free space magnetic moment of TM atoms by generating a significant amount of magnetic moments on Zn and Te sites,[65] as is depicted in Table
In addition, the exchange constants, N
0
α and N
0
β have also been calculated, which play a significant role in elucidating the real origin of ferromagnetism in DMSs. The Hamiltonian 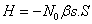
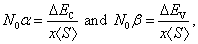 | (3) |
3.3. Optical properties
The wide range of potential applications of Zn-based II–VI DMSs in the field of optoelectronics indicates that they may replace the silicon-based semiconductor technology. The calculated complex dielectric constant 

 | Fig. 4. (color online) (a) The real ε
1(ω) and imaginary part ε
2(ω) of the dielectric constant, (b) refractive index n(ω) and extension coefficient k(ω) plotted against incident energy for Zn0.9375TM0.0625Te (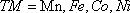
|
The imaginary part ε
2(ω) of the complex dielectric constant is a key parameter to decide a suitable material for a suitable optoelectronic device. The ε
2(ω), as plotted in Fig.
The refractive index n(ω) and the extinction coefficient k(ω) have also been computed and have depicted a behavior similar to ε
1(ω) and ε
2(ω), respectively (see Fig. 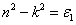
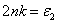
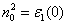


4. Conclusion
The structural, magnetic and optical properties of 

Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] | |
| [33] | |
| [34] | |
| [35] | |
| [36] | |
| [37] | |
| [38] | |
| [39] | |
| [40] | |
| [41] | |
| [42] | |
| [43] | |
| [44] | |
| [45] | |
| [46] | |
| [47] | |
| [48] | |
| [49] | |
| [50] | |
| [51] | |
| [52] | |
| [53] | |
| [54] | |
| [55] | |
| [56] | |
| [57] | |
| [58] | |
| [59] | |
| [60] | |
| [61] | |
| [62] | |
| [63] | |
| [64] | |
| [65] | |
| [66] | |
| [67] | |
| [68] | |
| [69] | |
| [70] | |
| [71] | |
| [72] |