



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Tracking molecular structure deformation of nitrobenzene and its torsion–vibration coupling by intense pumping CARS
[Wang Chang1, 2, Wu Hong-Lin1, Song Yun-Fei3, He Xing1, Yang Yan-Qiang1, 3, †,  , Tan Duo-Wang3]
, Tan Duo-Wang3]
, Tan Duo-Wang3]
|
|


† Corresponding author. E-mail:
Project supported by the National Natural Science Foundation of China (Grant Nos. 21173063 and 21203047), the Foundation of Heilongjiang Bayi Agricultural University, China (Grant No. XZR2014-16), NSAF (Grant No. U1330106), and the Special Research Project of National Key Laboratory of Shock Wave and Detonation Physics, Institute of Fluid Physics, China Academy of Engineering Physics (Grant No. 2012-S-07).
The structural deformation induced by intense laser field of liquid nitrobenzene (NB) molecule, a typical molecule with restricting internal rotation, is tracked by time- and frequency-resolved coherent anti-Stokes. Raman spectroscopy (CARS) technique with an intense pump laser. The CARS spectra of liquid NB show that the NO2 torsional mode couples with the NO2 symmetric stretching mode, and the NB molecule undergoes ultrafast structural deformation with a relaxation time of 265 fs. The frequency of NO2 torsional mode in liquid NB (42 cm−1) at room temperature is found from the sum and difference combination bands involving the NO2 symmetric stretching mode and torsional mode in time- and frequency-resolved CARS spectra.
Laser spectroscopic techniques have been successfully applied to chemistry and biology, providing important information about and insight into molecules and their environment.[1] When the intensity of nonresonant laser reaches 1011–1012 W/cm2, the torque generated by the interaction between nonresonant laser fields and the induced dipole moment of the molecule can provide an “instantaneous” kick and induce molecules to be aligned along the direction of the laser field and deformed.[2] One of the most widely used methods for confirming molecular deformation under intense laser fields is the Coulomb explosion (CE) method. In the CE method, the ultrafast nonresonant intense field causes multiple ionization of the parent molecule. The information of molecular structure deformation prior to explosion is given by mapping the distribution of the fragment ions. Although this methodology is very powerful, it cannot monitor the dynamics of molecular structure evolution after the molecular structure deformation.[3] The molecular dynamics prior to explosion can only be indirectly deduced from the distribution of fragment ions. Thus the CE method is destructive. Moreover, it requires the sample to have simple structure, such as CO2 and CS2, and to be gas phase.[4]
Recently, the modified coherent anti-Stokes Raman spectroscopy (CARS) technique with an intense nonresonant femtosecond pump laser (1011–1012 W/cm2) is used to drive the structural deformation of liquid methyl iodide (CH3I) and nitromethane (CH3NO2) molecules and track their relaxation process simultaneously.[5,6] The CARS is a nonlinear four-wave mixing process, and a Raman active vibrational mode is coherently driven when the energy difference between a pump laser and a Stokes laser is resonant with it.[7] CARS spectroscopy has been applied to investigate the ultrafast dynamics of molecules in the condensed phase.[8] This technique is essentially nondestructive and nonintrusive.[6] CH3I is the type of molecule without internal rotation, and CH3NO2 is the type of molecule with free internal rotation. These two situations have been discussed. Nitrobenzene (NB) has the simplest structure in aromatic nitro compounds, which are widely known as energetic materials.[9] NB molecule consists of NO2 group and phenyl rings. It has internal rotation and high barrier to internal rotation of 12.3 kJ/mol.[10] So NB represents the type of molecule with restricting internal rotation. Usually, the torsional vibration was observed in Raman experiments only at very low temperature.[11] The exact value of torsional mode of liquid NB has not been known.
In the present work, we perform the intense pumping and time- and frequency-resolved CARS on liquid NB and successfully track the structural deformation induced by intense nonresonant femtosecond laser pulse in real time. We also get the value of torsional mode of NO2 group of liquid NB (42 cm−1) from the process of molecular deformation.
In this time- and frequency-resolved CARS experiment, a high intensity (∼1012 W/cm2) pump pulse is used to provide the intense field to drive the torsional motion of NB molecules in the liquid phase and it is due to the effect of Impulsive Raman of femtosecond laser. A brief description is given here, and the other details of our experimental apparatus have been described in Ref. [12]. A 1 kHz regenerative amplifier was used to create the femtosecond pulse with a center wavelength of 800 nm, single pulse energy of 1.0 mJ, and pulse duration of 110 fs. The laser pulse was divided into three parts by two beam splitters (BS). Two of them were used as the pump and probe pulses. The third one produced an ultra-broadband white-light continuum (WLC) as the Stokes pulse by passing a 4-mm-thick Al2O3 crystal.[12] The WLC has an ultra-broadband spectral profile, ranging from 400 nm to 1100 nm. The pump light was focused by a 175 mm focal length lens, and the intensity of pump laser pulse was ∼1012 W/cm2. The intensities of Stokes and probe were set much lower than that of the pump pulse. Two optical delay lines were employed. One was used to produce the temporal overlap of the pump and Stokes pulse. Another one was used to provide the variable time delay for the probe pulse. The zero delay time was defined as the time when the pump and probe pulses overlap. The folded BoxCARS configuration with properly chosen angles between the pulses, defined by the four-wave-mixing (FWM) phase-matching condition, was employed in order to separate the signal from the incoming beams.[13] All three beams were sent through a lens focused onto the sample. The CARS signal generating in a direction
Time- and frequency-resolved CARS spectra of liquid NB are shown in Fig.
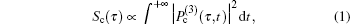
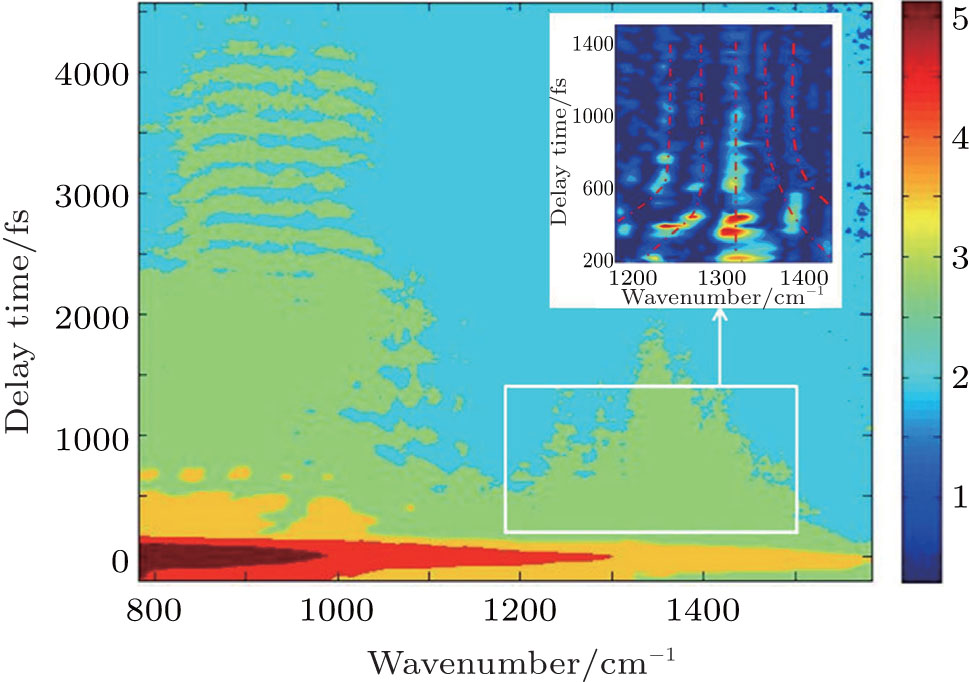 | Fig. 1. Logarithmic contour plot of the CARS signal intensity obtained from the sample of liquid NB. The two axes represent delay time and Raman vibrational frequency values, while contours represent the intensity of CARS signal. The inset shows the data in the frequency range of 1200–1500 cm−1 has been processed by subtracting their lower envelope. |
The lower frequency band shows the obvious quantum beating phenomena and decays within about 4.5 ps. It is due to the vibrational modes which are coherently excited by the femtosecond laser pulses with large spectral bandwidth. According to the Raman spectrum of NB, NON bending (852 cm−1), C–C–C trigonal bending (1003 cm−1), C–H bending (1021 cm−1), C–H bending (1074 cm−1), C–N stretching (1107 cm−1), and C–H bending (1172/1162 cm−1) mode can be found in this frequency range.[18] In order to obtain the accurate information about the dynamics, the temporal evolution of CARS signal at the spectral position of 988 cm−1, corresponding to the arithmetic mean wavenumber of the intense vibrational modes 852, 1003, and 1107 cm−1, is extracted. The time-dependent CARS signal at 988 cm−1 is shown in Fig.
 | Fig. 2. Logarithmic CARS signal as a function of probe pulse delay time at 988 cm−1. The inset shows the corresponding FFT spectrum of the beating component. |
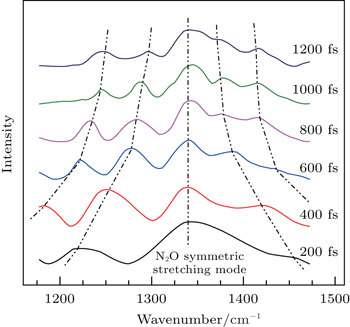
In the case of higher frequency band centered at 1345 cm−1 (NO2 symmetric stretching mode), no FFT analysis was necessary as there was only one prominent Raman mode in this frequency range and the CARS signal decays exponentially. For a single vibrational mode, the time-dependent intensity is fit by[19]

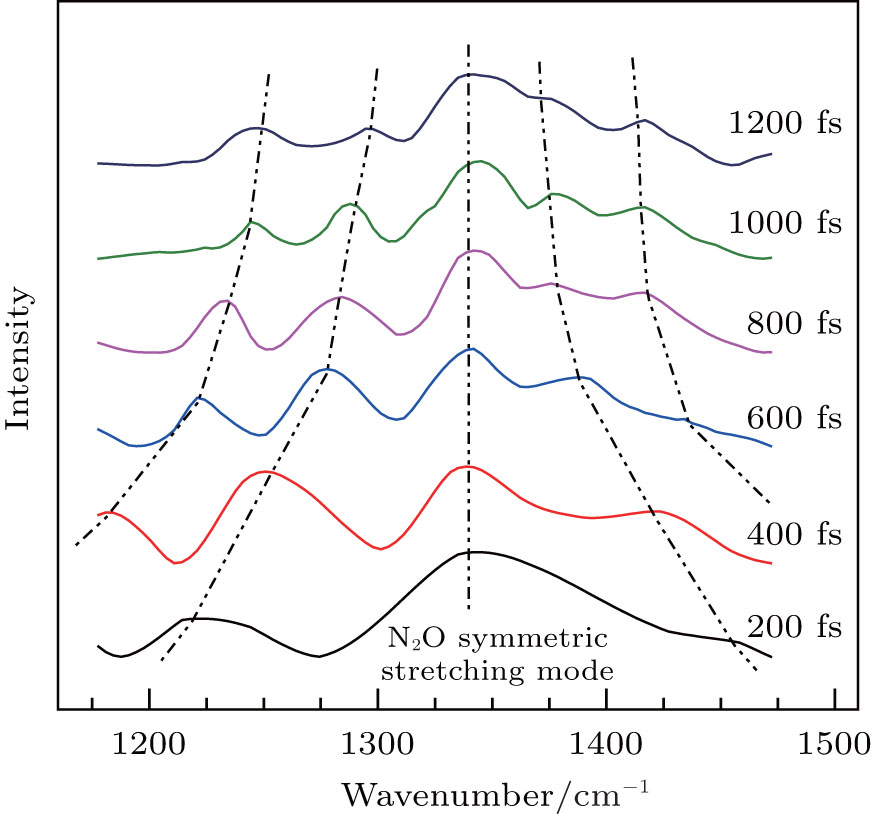 | Fig. 3. CARS spectra of symmetric stretching mode (1345 cm−1) of liquid NB as a function of the time delay. Spectra obtained at the different delay time (200, 400, 600, 800, 1000, and 1200 fs). The curves (dotted line) indicate the converging trend of peaks. |
The nonresonant laser pulse of intensity 1011–1012 W/cm2 can create alignment and deformation of molecules, such as CO2 and CS2.[4] It is due to the fact that the intense pump laser provides an “instantaneous” kick to induce deformation of molecular geometrical structure. The alternating electric field
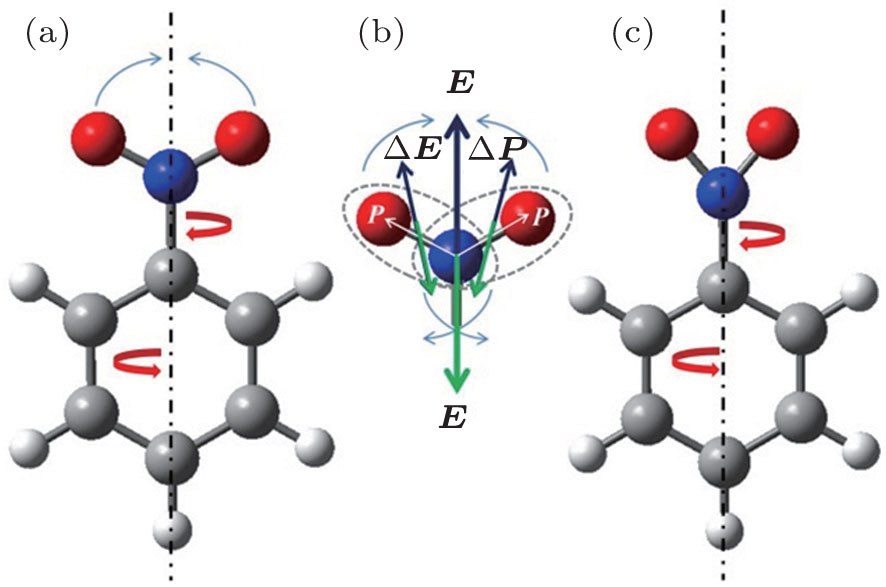 | Fig. 4. Structure of equilibrium conformation (a) and deformed shape (c) of NO2 group in the ball-stick model of NB molecule. Schematic of mechanism for molecular deformation of NO2 group induced by intense laser field (b). The blue, red, gray and white tubes refer to nitrogen, oxygen, carbon and hydrogen atoms, respectively. |
NB molecule has been considered as an internal rotation case with high barrier. When the barrier is so high that there is insufficient energy available under normal experimental conditions for this to be surmounted, the molecule undergoes a torsional vibration. It is the restricted rotation of a part of a molecule with respect to the molecular skeleton.[20] The frequency of torsional mode of NO2 can be denoted by νTors(NO2). It describes the restricted rotation of NO2 group of NB about the molecular axis of C–N bond, and is denoted by the red arrows in Figs.
The value of νTors(NO2) decreases with the delay time due to the influence caused by the structure deformation of NB molecules. At the zero delay time, the ultrafast structural deformation of NO2 group from the equilibrium conformation (Fig.
The relaxation of frequency of torsional mode is analyzed. This indicates the relaxing process of NB molecular structural deformation. The empirical exponential function is used to fit the frequency of the torsional mode at different delay times as follows:[6]
 | Fig. 5. Decay of the frequency of the torsional mode (black scatter) and the fitted curve (red solid curve). |
The frequency value of NO2 torsional mode of liquid NB at stable molecular structure is assigned at 42 cm−1 in our experiment. A microwave intensity measurement gives a value of 50±15 cm−1 for the fundamental torsional frequency of nitrobenzene molecule in the gas phase.[22] The far infrared spectrum shows a broad low frequency band value between 20 and 70 cm−1 in liquid NB.[23] The exact value of torsional mode of liquid NB has not been known. Clearly resolved torsional mode of NB is not observed in traditional Raman spectrum at room temperature. Usually the torsion of molecule is the lowest frequency mode below 100 cm−1, and torsional excitation would undergo strong damping when the temperature is increased. At higher temperature, the residence time becomes lower than the characteristic time (1 ps) linked to phonon bands, thus the torsional transitions are not observed in Raman experiments only at room temperature.
In our experiment, the intense laser pulses can induce torsional motion in a molecule and make the torsional mode with several vibrational excited state sufficiently populated at room temperature. While the torsional mode is coupling with the NO2 symmetric stretching mode, the pump and Stokes pulse excite coupling to make the residence time of coupling longer. Also CARS technique is the background-free nature of signal detection by the phase-matching conditions associated with the third-order polarization.[24] So the analysis of sum and difference combination bands involving the NO2 symmetric stretching mode provides accurate value of torsional mode.
In conclusion, intense pumping and time- and frequency-resolved CARS technique successfully monitors the structural deformation of NB molecules. The evolution of sum and difference combination bands involving NO2 symmetric stretching modes vs(NO2) and NO2 torsional mode vTors(NO2) is observed. The analysis of combination bands provides information of ultrafast structural deformation of NO2 groups induced by the intense femtosecond laser field. The relaxation time is about 265 fs. Meanwhile, the frequency value of torsional mode in liquid NB can be obtained from the CARS spectra. The similar experiment are performed on the samples of three different types of molecules which are molecule without internal rotation (CH3I),[5] molecule with free internal rotation (CH3NO2),[6] and molecule with restricting internal rotation (C6H5NO2), respectively. These investigations show that the modified CARS with intense pump pulse is a feasible and effective method to investigate structural deformation of molecules in the liquid phase induced by intense nonresonant femtosecond laser field without complex experimental system.
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 | |
| 23 | |
| 24 |