Lu Peng-Fei, Wu Li-Yuan, Yang Yang, Wang Wei-Zheng, Zhang Chun-Fang, Yang Chuang-Hua, Su Rui, Chen Jun. Stable structure and optical properties of fused silica with NBOHC-E′ defect. Chinese Physics B, 2016, 25(8): 086801
Permissions
Stable structure and optical properties of fused silica with NBOHC-E′ defect
Lu Peng-Fei1, 2, †, , Wu Li-Yuan1, Yang Yang1, Wang Wei-Zheng1, Zhang Chun-Fang3, Yang Chuang-Hua4, Su Rui3, Chen Jun5
State Key Laboratory of Information Photonics and Optical Communications, Ministry of Education, Beijing University of Posts and Telecommunications (BUTP), Beijing 100876, China
State Key Laboratory of Functional Materials for Informatics, Shanghai Institute of Microsystem and Information Technology, Chinese Academy of Sciences, Shanghai 200050, China
Beijing Computational Science Research Center, Beijing 100084, China
School of Physics and Telecommunication Engineering, Shaanxi University of Technology, Hanzhong 723001, China
Institute of Applied Physics and Computational Mathematics, Beijing 100088, China
Project supported by the National Basic Research Program of China (Grant No. 2014CB643900), the Open Fund of IPOC (BUPT), the Open Program of State Key Laboratory of Functional Materials for Informatics, the National Natural Science Foundation for Theoretical Physics Special Fund “Cooperation Program” (Grant No. 11547039), and Shaanxi Provincial Institute of Scientific Research Plan Projects, China (Grant No. SLGKYQD2-05).
Abstract
Abstract
First-principles method is used to simulate the stable structure and optical properties of a 96-atom fused silica. The preferable structure of NBOHC-E′ (non-bridging oxygen hole center (NBOHC) and E′ center) pair defect is predicted to be located at 2.4 Å for the Si–O bond length. The quasi-particle G0W0 calculations are performed and an accurate band gap is obtained in order to calculate the optical absorption properties. With the stretching of the Si1–O1 bond, an obvious redshift can be observed in the absorption spectrum. In the case of NBOHC-E′ pair, the p-orbital DOS of Si1 atom will shift to the conduction band. Two obvious absorption peaks can be observed in the absorption spectrum. The calculation reproduced the peak positions of the well-known optical absorption bands.
Properties of fused silica have attracted attention in the past decade due to its potential technological applications, including microelectronics and optical transmission media. Basically, it is crucial to stabilize specific defects by controlling the thermochemical process if we are to better understand their structural and electronic properties. Whether they are generated by manufacturing, irradiation, or mechanical deformation, the defects capable of trapping charge in silica are responsible for degrading device performance, leading to threshold voltage shifts in metal-oxide-semiconductor transistors, actuation voltage changes in mechanical systems, and even attenuation in high power laser devices.[1,2] Two main paramagnetic defects, i.e., the non-bridging oxygen hole center (NBOHC) and E′ center where E′ refers to a three coordinate Si atom,[3] can be observed at room temperature in irradiated fused silica, which have been considered to dominate the light absorptions. As is well known, the NBOHC is created by breaking the Si–O bond and forming the NBOHC-E′ pair. Therefore, the NBOHC represents the oxygen dangling bond or non-bridging oxygen hole center with a structure of ≡Si–O·, where (≡) refers to bonds with three oxygen atoms and (·) denotes an unpaired electron.[4,5]
Extensive experimental and theoretical studies[6–16] have been devoted to point defects in silica due to their relevance in the optical transmission properties of SiO2-based electronic devices. Bakos et al.[7] reviewed the experiments that are used to support the identification of the NBOHC defect responsible for the optical absorption (OA) at 4.8 eV and photoluminescence (PL) bands at 1.9 eV. In Ref. [8], three OA peaks of NBOHC in SiO2 glass induced by low-temperature F2 laser-irradiation are evident: the most intense peak at 4.8 eV, a medium intensity peak at ∼ 6.8 eV, and a low-intensity peak at ∼ 2 eV. Benoit et al.[9] and Cannas et al.[10] presented the OA bands extended from visible (2.0 eV), to ultraviolet (UV) (4.8 eV) and deep ultraviolet (DUV) (6.8 eV) regions. In the theoretical aspect, Raghavachari et al.[11] studied the OA of a series of point defects in SiO2 cluster models by the time-dependent density functional response theory (TD-DFT). Suzuki et al.[12] calculated the electronic structures and the natures of optical transitions in oxygen dangling bond in silica glass. Saeta and Greene[13] discussed the kinetics of photoinduced NBOHC-E′ defect formation in high-purity silicas by femtosecond transient absorption spectroscopy in the visible and UV regions. For the typical feature of NBOHC-E′ pair defect, Giordano et al.[17] reported that the terminal Si–O bond in an NBO· defect of hydroxylated silica surface is close to 1.69 Å. Although several studies have focused on optical transitions mode and the OA properties, the prediction of the preferable structure with representative defects in fused silica remains a significant computational challenge.
In this paper, a series of ab initio calculations are carried out to reveal the preferable structure, electronic and optical properties of an NBOHC-E′ pair in fused silica. A typical model with the NBOHC-E′ pair is generated by using molecular dynamics (MD) simulation. The structural optimization is realized by using the generalized gradient approximation (GGA) with the framework of first-principles DFT.[18–20] The electronic and optical properties are investigated by using the Bethe–Salpeter equation (BSE) based on the quasi-particle G0W0 method[21] in order to improve the accuracy of the calculation. The rest of this paper is organized as follows. The computational methods and models are described in Section 2. Our results and discussion are presented in Section 3. Finally, some conclusions are drawn from the present study in Section 4.
2. Computational method and model construction
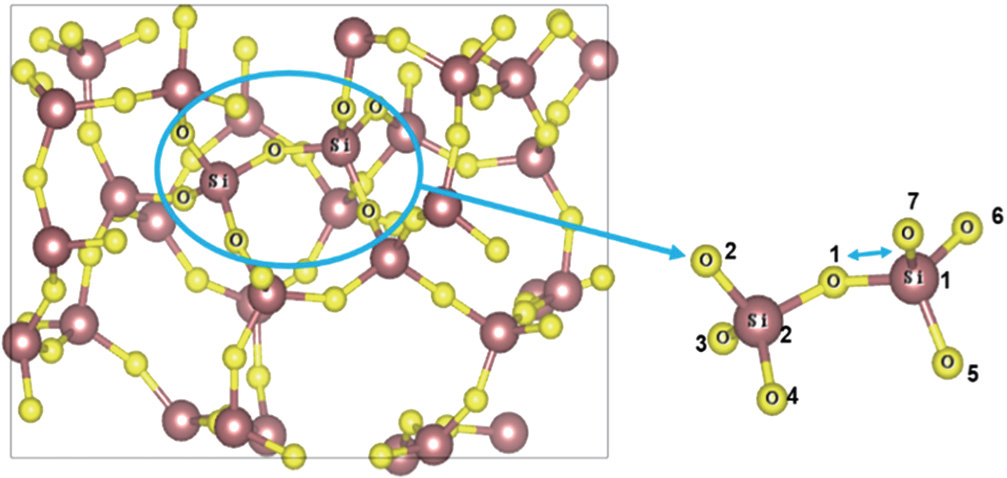
A 2×2×2 supercell containing 96 atoms is built and optimized with a force tolerance of 0.1 eV/Å. The size of supercell (∼ 10 Å) is large enough to neglect the interaction with its periodic images. A Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) based on the classical MD method[22] is used to construct the fused silica model, where a three-stage heating-cooling procedure is performed. At first, the 96-atom supercell system is heated for 50 ps under periodic boundary conditions (PBCs) with an initial temperature of 5000 K to achieve the condition of equilibrium; then, it is quenched to 500 K by using 9 chained quenching-equilibrating steps; finally, the system is cooled to 300 K and equilibrated for 50 ps. Subsequently, equilibration MD simulations are performed in time steps of 0.5 fs under the isothermal-isobaric (NPT) ensemble. The external pressure is set to be the air pressure under ambient conditions. The system temperature is controlled by using the Langevin thermostat. The simulated density for the supercell is found to be 2.2 g/cm3, which agrees well with the experimental result (2.2 g/cm3).[23] Figure 1 shows the 96-atom supercell of fused silica without defect.
Fig. 1. Perfect 96-atom supercell model for fused silica. The NBOHC-E′ pair will be generated from the extracted portion. Si is shown in brown, and O is in yellow. All the calculations are based on the supercell model.
First-principles simulations are performed in the framework of DFT implemented in Vienna ab initio simulation package (VASP).[24,25] The GGA exchange-correlation functional in the Perdew–Burke–Ernzerhof (PBE) form[26] is used to describe the electron correlation. The electron wave function is expanded in plane waves with a cutoff energy of 450 eV, which is tested to give converged results. The Brillouin-zone is sampled in the k space within the Monkhorst–Pack scheme by 2×2×2 mesh points and a small Gaussian broadening σ = 0.1 eV is used. Geometry minimizations are used in the single-point calculations to validate the geometry of the NBOHC-E′ pair. The optimization will be completed when the following conditions are satisfied (the maximum force on each atom achieves 0.01 eV/Å, and the maximum energy change between two ionic steps converges at 10− 5 eV). The electronic structure and optical properties of the NBOHC-E′ pair are obtained by solving the BSE based on the quasi-particle G0W0 calculations.
3. Results and discussions
3.1. Stable structure with NBOHC-E′ center
A typical NBOHC-E′ pair defect is generally created by breaking the Si–O–Si bond, which is caused by a bound electron-pair. The O1 is “knocked” out of the Si1–O1 network, and then trapped by the nearby atoms and forms a new ring. Therefore, a key problem is to predict the preferable Si1–O1 distance in order to represent the actual NBOHC-E′ defect. In Fig. 1, the perfect 96-atom supercell for fused silica is shown and part of the supercell which we select to create the defect is illustrated in the blue circle and this model is named Model A. In search for the stable structure, the Si1–O1 bond varies from 1.5 Å to 3.0 Å and the initial stable distance between Si1 and O1 is 1.6 Å after the structural optimization. The length of Si2–O1 bond shows a stable oscillation around 1.63±0.01 Å. Both of the singlet and triplet states are searched to obtain the structures of the ground and excited states. The excited state is gained by fixing the electron occupation on the up and down spin channels. The search of the sable state becomes seeking for the lowest saddle points of the minimum energy path (MEP).[27] The MEP often has one or more minima in addition to the minima at the initial and final states. These correspond to stable intermediate configurations.
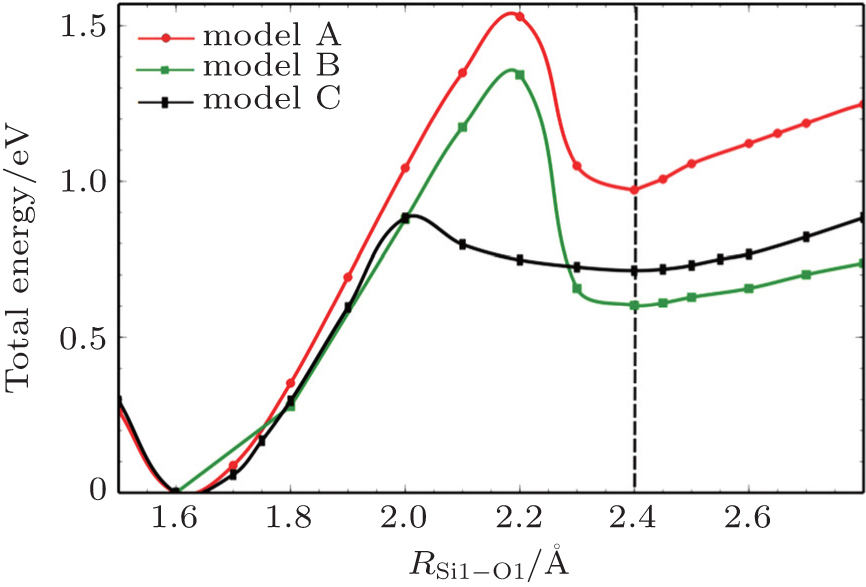
Figure 2 shows the energy curves for the singlet/triplet state in the supercell as the Si1–O bond length varies, where the black line is for the singlet state and the red one is for the triplet state. As can be seen in Fig. 2, for the singlet state, a stable structure has an Si1–O1 bond length of 1.6 Å. With the increase of Si1–O1 bond length, the total energy for the system increases nonlinearly. For the triplet state, the first stable structure is almost the same as that of the singlet state, and 1.6 Å is the perfect balance position of fused silica without defect. With the augment of the Si1–O1 bond length, the energy increases until a drop in potential energy surface appears at 2.2 Å for Si1–O1 bond length and then a shallow valley (the second stable structure) appears with the Si1–O1 bond fixed at 2.4 Å, which is found to be the preferable NBOHC-E′ structure. The energy barrier for the first transition between 1.6 Å and 2.2 Å is 1.53 eV, while for the second transition between 2.2 Å and 2.4 Å it is 0.52 eV.
Fig. 2. Total energies versus RSi1−O1 for singlet and triplet state.
To verify the feasibility of our prediction for the preferable NBOHC-E′ structure, two more models are constructed and the calculated preferable structures are shown in Fig. 3. Model B refers to the structure with 96-atom supercell just the same as model A, while the NBOHC-E′ pair is at a different location. Model C is defined as a 72-atom supercell of fused silica. These three models show the consistent results that 1.6 Å is the perfect balance position for defect free structure. The size effect of supercell will affect the position of transition state. However, the preferable state of NBOHC-E′ structures (2.4 Å of Si–O bond length) are almost identical for these three models.
Fig. 3. Predicted preferable NBOHC-E′ structures from different models. Model A refers to the Si1–O1 bond in Fig. 1. Model B represents a model with the defect at a different location from model A. Model C means a small supercell with 72 atoms. Total energy marked on the Y axis represents the energy relative to the energy value of 1.6 Å in model A.
3.2. Electronic structure
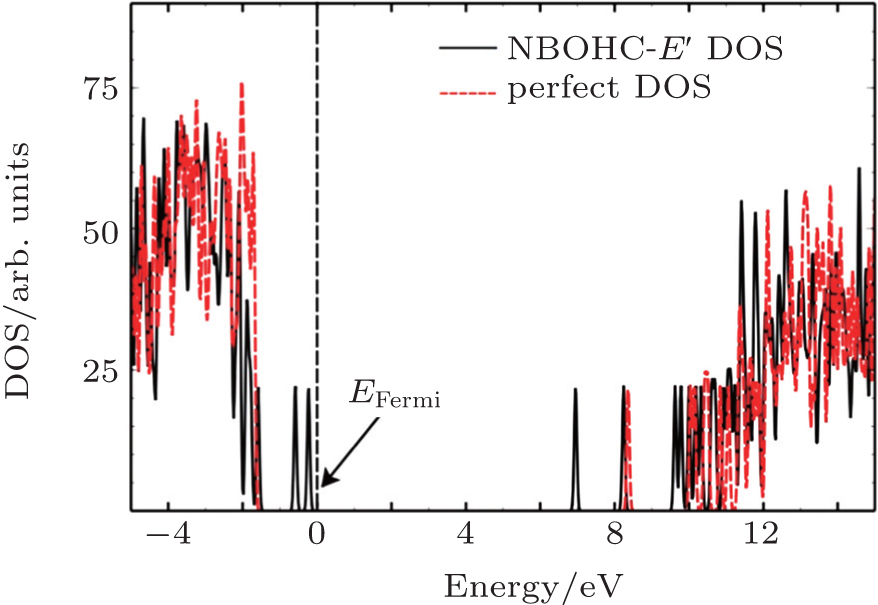
GGA-PBE functional in DFT is used to calculate the band structure of the fused silica model in Fig. 1. The calculated band gap is about 5 eV, which is lower than the experimental data of 9 eV.[28] As is well known that DFT sometimes underestimate certain results.[29] To improve the accuracy, quasi-particle energies of the system are estimated by using the G0W0 approximation. The main advantage is that the quasi-particle eigenvalues can be obtained as a first-order perturbation of a previous Kohn–Sham set of eigenvalues. The density of states (DOS) for the perfect model and that with the NBOHC-E′ defect are described in Fig. 4. The DOS of a perfect supercell is aligned with the Fermi energies of defect structures for comparison. The Fermi energy of defect structures is set to be zero. For perfect fused silica, our calculated band gap is 9.66 eV. For fused silica with the NBOHC-E′ structure, there is no obvious shrink of band gap. However, three main defect levels can be observed to be above the valence band, and below the conduction band. In the valence part of the DOS of the NBOHC-E′, two defect levels include the non-bonding 2p-orbitals of O and the low lying bonding orbitals are produced beyond the valence band maximum (VBM) with a gap of 0.36 eV. The unoccupied state is located at 6.9 eV in the conduction band. The formation of the NBOHC-E′ defect results in the breaking of the Si–O bond and introduces two occupied states near the Fermi level and an unoccupied defect state.
Fig. 4. Total DOSs of perfect supercell (red dashed line) and fused silica with NBOHC-E′ defect (black solid line).
To further depict the contribution of band structure, the partial densities of states (PDOSs) are introduced to explain the origins of these impurity states. The corresponding PDOSs of the Si atoms and the adjacent O atoms are plotted in Fig. 5. Figure 5(a) refers to p-orbital DOSs of Si1 and O1 compared with those for the perfect supercell, while figure 5(b) represents those of the NBOHC part. In Fig. 5(a), Si1 and O1 are from the fused silica with NBOHC-E′ structure; and Si1 (perfect) and O1 (perfect) are picked up from the perfect fused silica model. Near −1 eV, blue and red dotted lines overlap, however, green and black solid lines which represent the NBOHC-E′ defect do not (Fig. 5(b)). As the center atom of the E′ defect, the p-orbital of Si1 shifts to the conduction band. When the Si1–O1 bond is stretched to 2.4 Å, a clear overlap between the Si 3p and O 2p states can be observed, and will be responsible for the two occupied states near the Fermi level and the unoccupied defect state in the conduction band. The Si1–O1 bond is broken with the charge transferring from the bonding orbitals to the anti-bonding orbitals. In Fig. 5(b), the 3p state of three-coordination Si2 atom will contribute to the 6.9 eV defect state. These impurity states result in a little reduction of the SiO2 band gap, which is in accordance with Fig. 4, and also are the transition states to assist photon absorption.
Fig. 5. PDOSs of fused silica. (a) p-orbital of O1 (black) and Si1 (green) in the defect model compared with O1 (red) and Si1 (light blue) in the perfect supercell. (b) p-orbital of O2 (pink), O4 (black), and Si2 (blue) in the defect model.
3.3. Charge distribution and bonding properties
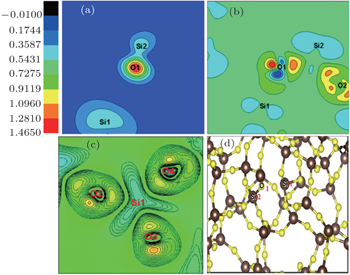
In order to understand the forming mechanism of the defect states, the partial charge density for the defect states are shown in Fig. 6. In Fig. 6(a), where the NBOHC-E′ structure is formed, the charge of O1 is mainly shared with Si2, and Si1 is converted into a three-coordinate atom. Figures 6(b) and 6(c) show the differential charge density of the NBOHC-E′ structure with the Si1–O1 bond fixed at 2.4 Å. In Fig. 6(b), the partial charge transfers to the side near the non-bridging oxygen atom. There are two obvious charge accumulations near the non-bridging oxygen atom. In Fig. 6(c), Si atom bonds with three O atoms, forming the space structure of E′. The three angles of O–Si–O are 116.2°, 104.5°, and 106.8°, respectively and the three distances of the Si–O bond are 1.672 Å, 1.674 Å, and 1.653 Å. It is noted that there exists a clear covalent bond between Si and O atoms.
Fig. 6. Partial and differential charge density calculations for the NBOHC-E′ structure. Panel (a) shows the partial charge density for the defect states in the Si1–O1–Si2 plane, panel (b) represents the differential charge density of the NBOHC-E′ structure, panel (c) displays the differential charge density of the plane centered on Si1 with E′, and panel (d) exhibits the optimized fused silica model with the NBOHC-E′ pair defect.
3.4. Optical properties
Based on the G0W0 calculations, optical properties of both defect and perfect structures are predicted by solving the BSE in the Tamm–Dankoff approximation. The OA coefficient is directly related to the imaginary part of the dielectric function.
Figure 7 shows the average dielectric imaginary functions of the perfect structure and the NBOHC-E′ structure (2.2 Å and 2.4 Å) based on non-spin approaches. The absorption edge is mainly from the interband transitions between the top of the valence band and the bottom of the conduction band. Two obvious in-gap OA peaks can be found at 2.0 eV and 4.5 eV for NBOHC-E′ models with a Si1–O1 bond length of about 2.4 Å compared with the perfect supercell as shown in Fig. 7. The first absorption peak may be induced by the self-trapped excitions localized at the broken Si1–O1 bond, and the second one may come from the electron transition between the non-bonding 2p orbitals of the non-bridging/bridging oxygen and the unoccupied defect state. Although there is a minor difference, the theoretical spectra obtained are close to the previous study for the amorphous SiO2 case.[30–33] Pacchioni et al.[30] found that the optical absorption and luminescence spectra of γ-irradiated wet silica can explain the main optical properties of NBOHC: the absorption/excitation bands at 4.8 eV and 1.97 eV, and the photoluminescence band at 1.9 eV. David Waroquiers[34] deemed that this discrepancy is thus likely to be attributed to the under-converged k-point grid. The missing spectral weight is expected to be recovered by including more than one k-point in the electron-hole basis set which in turn could reconcile the theoretical position of the peak with the experimental one. Figure 7 also indicates that the ZZ direction plays a dominant role in the OA spectrums.
Fig. 7. Different variations of ɛimag with energy in the three directions of three configurations for different Si1–O1 bond lengths.
4. Conclusions
In this work, the classical MD method is used to obtain a stable structure and optical properties for the 96-atom supercell of fused silica. The formation of the NBOHC-E′ center is found by stretching the Si1–O1 bond based on first-principles methods and the preferable NBOHC-E′ defect is found to have a Si1–O1 bond length of about 2.4 Å. The electronic and optical properties are studied by using the G0W0/BSE method. Compared with the perfect model, two obvious OA peaks of the NBOHC-E′ pair are found and show a redshift with the increase of Si1–O1 distance. Our results are of significance for comprehensively analyzing the inherent mechanism of the NBOHC-E′ defect which could be induced by high-power laser. This work can provide technique support for improving laser induced damage performance of fused silica, which is of significance for enhancing the working life of a laser transmission device.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 , Wu Li-Yuan1, Yang Yang1, Wang Wei-Zheng1, Zhang Chun-Fang3, Yang Chuang-Hua4, Su Rui3, Chen Jun5]
, Wu Li-Yuan1, Yang Yang1, Wang Wei-Zheng1, Zhang Chun-Fang3, Yang Chuang-Hua4, Su Rui3, Chen Jun5]