





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Molecular dynamics study of anisotropic growth of silicon
[Zhou Naigen1, Liu Bo1, Zhang Chi1, Li Ke1, Zhou Lang1, 2, †,  ]
]
]
|
|


† Corresponding author. E-mail:
Project supported by the National Natural Science Foundation of China (Grant Nos. 51361022, 51561022, and 61464007) and the Natural Science Foundation of Jiangxi Province, China (Grant No. 20151BAB206001).
Based on the Tersoff potential, molecular dynamics simulations have been performed to investigate the kinetic coefficients and growth velocities of Si (100), (110), (111), and (112) planes. The sequences of the kinetic coefficients and growth velocities are μ(100) > μ(110) > μ(112) > μ(111) and v(100) > v(110) > v(112) > v(111), respectively, which are not consistent with the sequences of the interface energies, interplanar spacings, and melting points of the four planes. However, they agree well with the sequences of the distributions and diffusion coefficients of the melting atoms near the solid–liquid interfaces. It indicates that the atomic distributions and diffusion coefficients affected by the crystal orientations determine the anisotropic growth of silicon. The formation of stacking fault structure will further decrease the growth velocity of the Si (111) plane.
Multicrystalline silicon (mc-Si) solar cells have been occupying the mainstream photovoltaic market because of their low production cost[1] and high stability.[2] However, the conversion efficiency of mc-Si solar cells is lower than that of single crystalline silicon solar cells. In order to improve the conversion efficiency of mc-Si solar cells, the mc-Si ingot has experienced three typical development stages in industry: common mc-Si, mono-like silicon, and high efficiency mc-Si. They focus on the techniques of lowering the dislocations density, controlling the crystal orientations and the sizes of the grains in the mc-Si ingot,[3–6] respectively. Anisotropic growth plays an important role in all these techniques.
There is little experimental literature about the anisotropic growth of Si crystal owing to the difficulty in experiment. Fujiwura[7] studied the competitive growth of the (100) to (111) planes of Si by building a bicrystal artificially, and observed the anisotropic growth at a high undercooling temperature. They have also found different morphological transformations at different solid–liquid interfaces by in situ observation, and proposed equations for calculating the growth velocity of Si ⟨112⟩ and ⟨110⟩ dendrites.[8–10]
The anisotropic growth is also a fundamental issue for crystal growth from melt. It has been extensively studied in the field of metal solidification. The anisotropic growth of crystal is mainly described by the kinetic coefficient μ, a very important parameter describing the movement of the solid–liquid interface. Many results showed that the kinetic coefficients of different growth planes in FCC metals[11–15] obey μ(100) > μ(110) > μ(111). Various theories were put forward to explain the above relationship. The typical theories consider the difference of interface energy,[16–20] interplanar spacing,[21] and formation of stacking fault.[22] Although Apte and Zeng[16] have obtained the (100), (110), and (111) solid–liquid interface energies of silicon by molecular dynamics, there is little in the literature studying the kinetic coefficient μ of the different growth plane of Si. In this paper, we calculate the melting points of different planes in order to set the undercoolings for them, and then investigate the kinetic coefficients and the crystal growth velocities of the different planes. Finally, the atomic distribution and the diffusion coefficient of the atoms near the solid–liquid interface are investigated to explain the anisotropic growth of silicon. This paper is to supply a reference for improving the quality of mc-Si and the conversion efficiency.
Figure
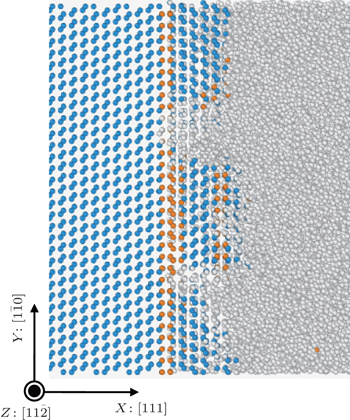
 | Fig. 1. Crystal melting and growth model of silicon (100) plane. |
The inter-atomic forces are calculated with the Tersoff potential,[23] which has been shown to be appropriate for modeling the transition from melt to crystal of silicon.[24] The Tersoff potential based on the concept of bond order in quantum mechanics was brought by Tersoff in 1986, and has been revised twice in 1988 and 1989, forming three editions T1,[25] T2,[26] and T3.[23] The Tersoff potential can well describe the diamond structure and the non-positive body structure. The formulas of the T3 potential are shown as follows:



Newton’s equations of motion are integrated with the velocity form of the Verlet algorithm using a time step of 0.001 ps. The simulation is performed in an NPT ensemble with periodic boundary conditions on three directions.
The Verlet algorithm[27] is the most commonly used algorithm in molecular dynamics. This algorithm is based on the third-order Taylor expansions for positions


Add these two expressions, and ignore the high order term, we have
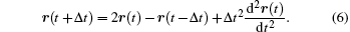
That is the basic expression of the Verlet algorithm.
The crystal growth velocity is calculated according to the variation of the number of crystal atoms (Nc).[28] To determine whether an atom belongs to the crystal or not, we calculate the number of its “neighbors”, which are the atoms within a sphere with a radius of the average distance of the first and the second nearest neighbor distances. If the number of neighbor atoms is equal to that of the first nearest neighbors in the corresponding perfect crystal, the atom at the center of the sphere is considered as a crystal atom, and vice versa. After adding up the crystal atoms in the system, the crystal growth or melting velocity can be calculated by the slope of the curve of crystal atom number versus time, and the formula is as follows:
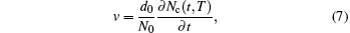
The melting point is the temperature at which melting or solidification of bulk materials occurs. The micro-process of melting always starts from the crystal defects in the surface, such as grain boundaries and dislocations. For a single crystal, the melting points of different planes should be different and will be a little higher than that of polycrystalline due to a lack of grain boundaries. We calculate the melting points of the Si (100), (110), (111), and (112) planes using the method of backward induction of melting velocity by molecular dynamics.[29] Firstly, the system is melted at different temperatures, and the melting velocities are calculated according to Eq. (
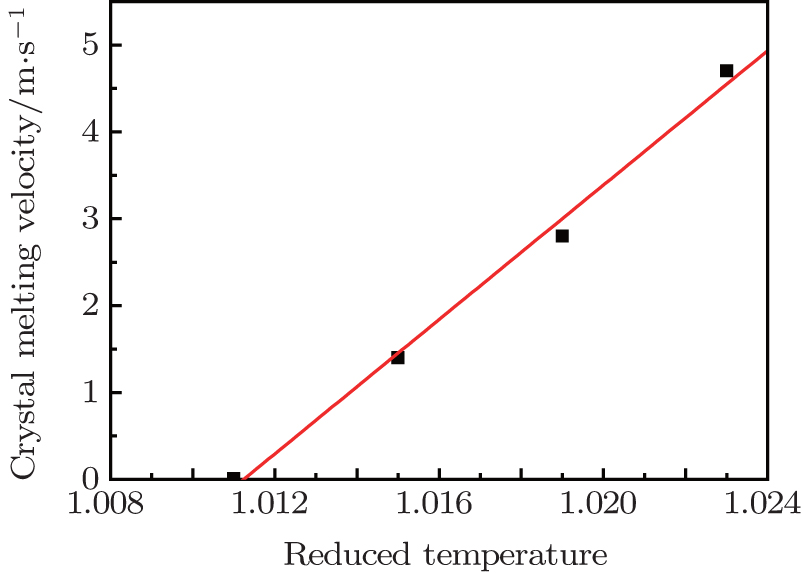 | Fig. 2. Crystal melting velocity of silicon (100) plane at reduced temperature. |
We extrapolate the curve of the melting velocity to zero, i.e., the cross point with the temperature axis, which gives the melting point of that plane. The melting points of Si (100), (110), (111), and (112) planes are obtained. The descending order of them is (100), (110), (111), and (112) planes. Table
| Table 1. Melting points of different planes of silicon. The value is relative to that of the (112) plane. . |
The kinetic coefficient is calculated by the free solidification method.[30] The simulations of the crystal growth along different planes have been performed at different undercoolings, which is set according to the melting point of each plane shown in Table
 | Fig. 3. Growth velocities of different planes of silicon at different undercoolings. |
The crystal growth velocities increase linearly with the increase of undercooling for the (100), (110), and (112) planes, and almost linearly for the (111) plane. The slopes of the fitting lines, the kinetic coefficients, are different. The corresponding values are μ(100) = 0.031 m·s−1 · K−1, μ(110) = 0.029 m·s−1 · K−1, μ(112) = 0.028 m·s−1 · K−1, and μ(111) = 0.0050 m · s−1 · K−1. The sequence is μ(100) > μ(110) > μ(112) > μ(111), which is very similar to that of the FCC metal,[11–15,29,31] μ(100) > μ(110) > μ(111). There are few studies on the (112) plane in metal. Here we pay attention to this plane because the dislocations easily emerge along it.
Anisotropic growth of silicon at different temperatures has also been investigated. The reduced temperatures (relative to the melting point of the (112) plane) are set here as 0.81, 0.85, 0.89, 0.93, and 0.97, respectively. The growth velocities are calculated according to the variation of the number of crystal atoms as described in the previous section. The results are shown in Fig.
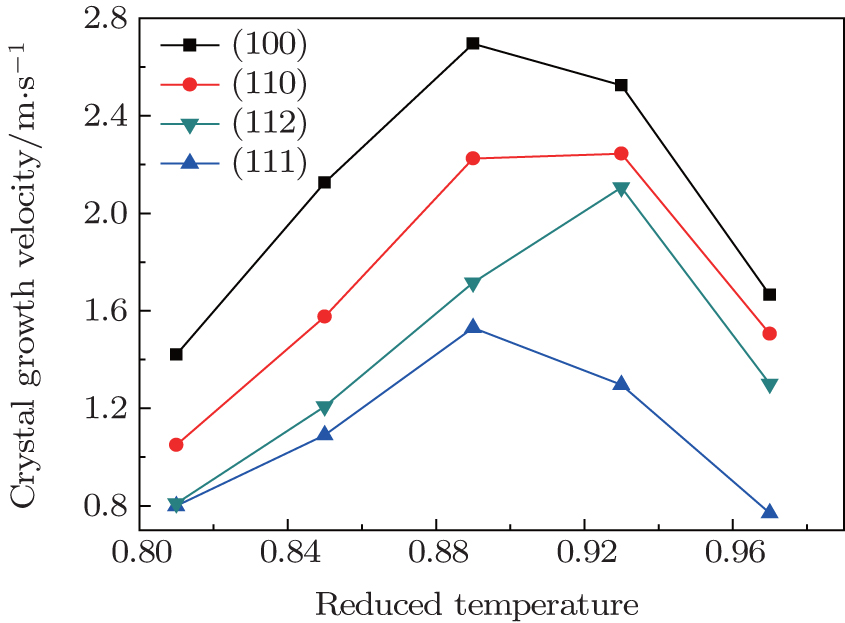 | Fig. 4. Crystal growth velocities of different planes as a function of the temperature. |
The difference of interfacial energy,[16] interplanar spacing,[21] and the formation of stacking fault[23] are always used to interpret the anisotropic growth. Table
| Table 2. The relevant parameters of Si (100), (110), (112), and (111) planes. . |
The interplanar spacing interpretation comes from the BGJ growth model.[22] The main point of this model is that the growth is governed by the collisions of the melting atoms with the growth interface. If the time of growing one layer is close for each plane, the interplanar spacing will play an important role in the growth. A plane with wider interplanar spacing will grow faster. Several studies on metals[11–15,29,31] have shown great agreement with this theory. For example, Hoyt[11] found that the ratio μ(100)/μ(110) is approximately equal to the ratio of the interplanar spacings of the (100) and (110) planes. As shown in Table
We investigate the influence of the orientation on the structure and the atomic movement of Si melt. Figure
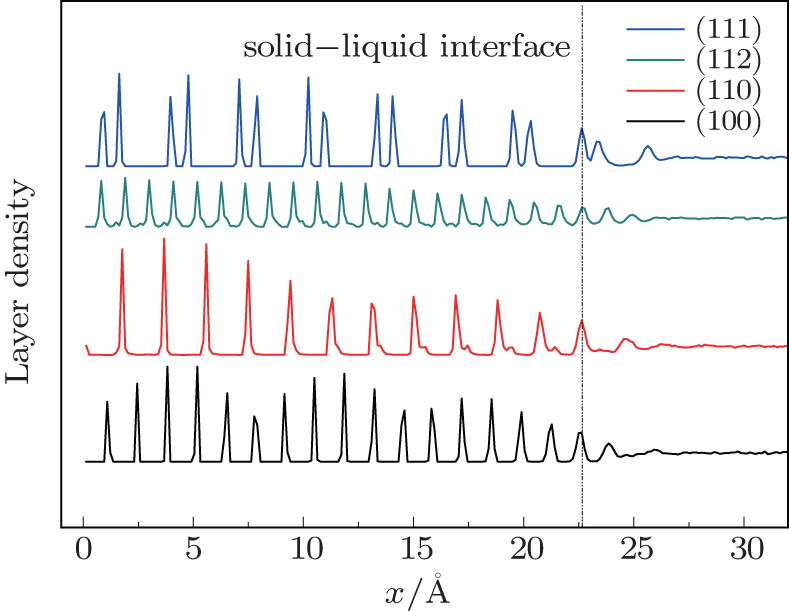 | Fig. 5. Layer densities along different orientations during the growth. |
We further calculate the diffusion coefficients of the melting atoms near the solid–liquid interfaces at the reduced temperature of 0.99 by the MSD method.[33] The results are shown in Fig.
 | Fig. 6. Diffusion coefficients near the solid–liquid interfaces at reduced temperature of 0.99. |
The kinetic coefficient and diffusion coefficient of the Si (111) plane are much lower than those of the other three planes as shown in Figs. 
 | Fig. 7. The  |
The relative melting point, kinetic coefficient, and growth velocity of Si (100), (110), (111), and (112) planes have been investigated by molecular dynamics. It is found that the melting points of the four planes are close. The melting points of Si (100), (110), (111) planes are 24 K, 15 K, and 13 K higher than that of the Si (112) plane, respectively. The sequences of the kinetic coefficients and the growth velocities are μ(100) > μ(110) > μ(112) > μ(111) and v(100) > v(110) > v(112) > v(111).
The analysis of the interface energy or interplanar spacing fails to interpret the anisotropic growth of Si, though it succeeds for metals. The distribution and diffusion coefficient of the melting atoms near the interface are affected by the crystal orientation. Their sequences agree well with the sequences of the kinetic coefficient and growth velocity, which can explain the anisotropic growth of Si. That is, the crystal orientations affect the distributions of the melt atoms near the solid–liquid interfaces, and then affect the diffusion coefficients of these atoms, which finally result in the anisotropic growth of the Si crystal.
The stacking fault structure can easily form due to the close-packed structure of the Si (111) plane. The forming of the stacking fault structure is a process of competitive growth between FCC sites and HCP sites, which will decrease the growth velocity of the Si (111) plane. It leads to the kinetic coefficient and growth velocity of Si (111) being much lower than those of the other three growth planes.
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 | |
| 23 | |
| 24 | |
| 25 | |
| 26 | |
| 27 | |
| 28 | |
| 29 | |
| 30 | |
| 31 | |
| 32 | |
| 33 |