{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Amyloid- β peptide aggregation and the influence of carbon nanoparticles
[Xi Wen-Hui , Wei Guang-Hong †,  ]
]
]
|
|


† Corresponding author. E-mail:
Project supported by the National Natural Science Foundation of China (Grant Nos. 11274075 and 91227102).
Soluble peptides or proteins can self-aggregate into insoluble, ordered amyloid fibrils under appropriate conditions. These amyloid aggregates are the hallmarks of several human diseases ranging from neurodegenerative disorders to systemic amyloidoses. In this review, we first introduce the common structural features of amyloid fibrils and the amyloid fibrillation kinetics determined from experimental studies. Then, we discuss the structural models of Alzheimer’s amyloid- β (A β ) fibrils derived from solid-state nuclear magnetic resonance spectroscopy. On the computational side, molecular dynamics simulations can provide atomic details of structures and the underlying oligomerization mechanisms. We finally summarize recent progress in atomistic simulation studies on the oligomerization of A β (including full-length A β and its fragments) and the influence of carbon nanoparticles.
Amyloid fibrillar aggregates, formed by the self-aggregation of peptides/proteins, not only have biological functions in certain organisms, [ 1 ] but also are one of the hallmarks of several human diseases ranging from neurodegenerative disorders to systemic amyloidoses. [ 2 ] More than 20 human diseases are associated with the pathological self-assembly of amyloid-forming peptides/proteins, such as Alzheimer’s Disease (AD), [ 3 ] Parkinson’s Disease (PD), [ 4 ] transmissible spongiform encephalopathy (TSE), [ 5 ] Huntington’s Disease, [ 6 ] and type II diabetes (T2DM). [ 7 ] For each disease, a specific protein is involved in amyloid fibril formation. Some of the aggregation-prone peptides/proteins have a well-defined three-dimensional (3D) structure in solution, while some are intrinsically disordered peptides/proteins (IDPs), [ 8 ] which typically consist of multiple charged residues and a few hydrophobic residues. Intensively-studied amyloidogenic IDPs include the amyloid- β (A β ) peptide (37–43 amino acids [aa], [ 9 ] associated with AD), tau protein (441 aa, [ 10 ] related to AD), the human islet amyloid polypeptide (37 aa, [ 11 ] associated with T2DB), and alpha-synuclein (140 aa, associated with PD). [ 12 ] Although the amyloidogenic proteins have different amino acid sequences and biophysical properties in solution, they all self-aggregate into thin and long amyloid fibrils, indicating their common structural feature [ 13 ] and aggregation mechanism. Although amyloid deposits are the hallmarks of many human diseases, a growing body of evidence suggests that the fibrillation process itself and the early formed small oligomers are also cytotoxic. [ 14 , 15 ] However, due to the transient nature of the oligomers, it is challenging to characterize their structures experimentally. As a complement to experimental methods, molecular dynamics (MD) simulations can provide atomic details of structures and the underlying oligomerization mechanism. In this review, we first introduce the common structural feature of amyloid fibrils and the amyloid fibrillation kinetics. We then discuss the structural models of A β fibrils derived from solid-state nuclear magnetic resonance (ss-NMR). Finally, we summarize recent progress in atomistic simulation studies on the oligomerization of A β (including full-length A β and its fragments) and the effects of carbon nanoparticles.
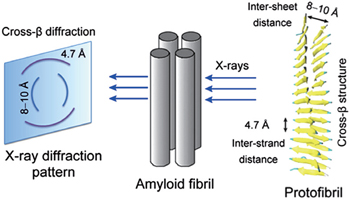
Based on the results of x-ray diffraction experiments, amyloid fibrils are known to be characterized by a cross- β structure, with the β -strands perpendicular to and the inter-strand hydrogen bonds parallel to the fibril axis [ 16 , 17 ] (for more details about the cross- β structure, see Fig.
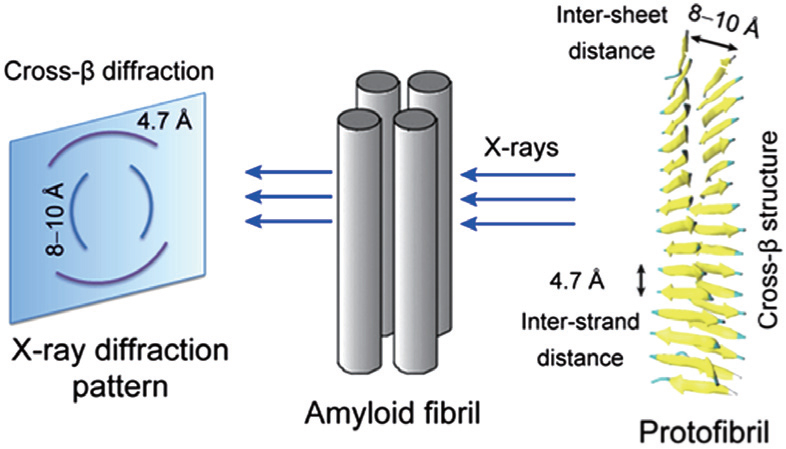 | Fig. 1. A diagram showing the x-ray diffraction pattern and cross- β structure feature of fibrils. The characteristic cross- β diffraction pattern is generated when x-rays are directed on amyloid fibrils, which consist of multiple protofibrils. The diffuse reflection at 4.7 Å spacing along the meridian (vertical) shows extended protein chains running approximately perpendicular to the fibril axis and spaced 4.7 Å apart. The increasingly diffuse reflection at 8–10 Å spacing along the equator (horizontal) shows that the extended chains are organized into sheets spaced 8–10 Å apart. The cross- β structure can be seen from the cartoon representation of a protofibril. |
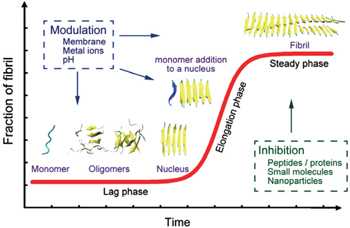
Understanding the self-aggregation process of amyloid peptides from soluble monomers/oligomers to insoluble amyloid fibrils is crucial for the investigation and treatment of amyloidosis. In vitro experimental studies of the kinetics of peptide aggregation have been carried out extensively. [ 20 – 22 ] The most well-accepted model is the nucleation-dependent polymerization model, where the fibrillation process produces a sigmoidal kinetic curve. [ 20 , 21 ] A schematic diagram of the fibrillation process is shown in Fig.
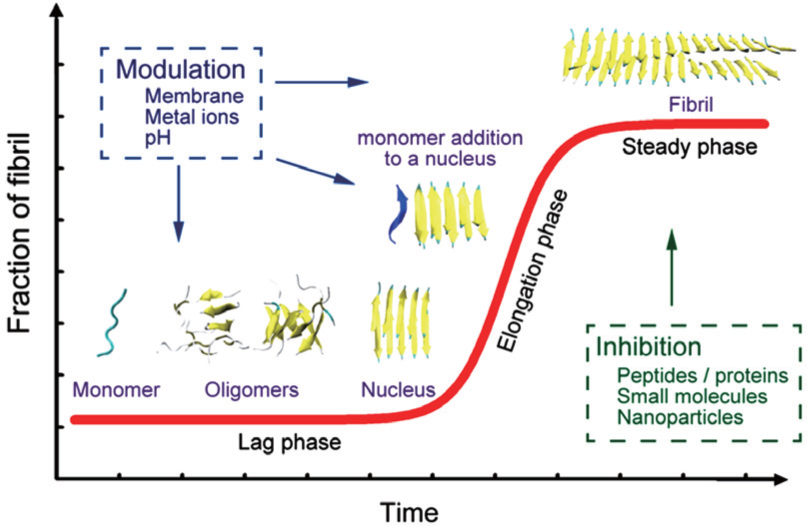 | Fig. 2. A schematic diagram showing the nucleation-dependent polymerization model of amyloid fibril formation. During the lag phase (nucleation), monomeric forms of the amyloid-forming peptides self-aggregate into soluble oligomers (including disordered oligomers, β -barrels, and bilayer β -sheets) and nuclei. During the elongation phase, the nucleus is extended rapidly and grows into fibrils. Then, fibrillation reaches a stage called the steady phase. Aggregation processes produce a sigmoidal kinetic curve (red) that is depicted in the figure. Many factors can influence this fibrillation process. [ 26 – 28 ] |
Alzheimer’s amyloid plaques consist mostly of 40- and 42-aa(A β 1–40 and A β 1–42) peptides. A β 1–42 is considered more toxic than A β 1–40, although the latter is more abundant in neurons. [ 29 ] Since 2000, based on structural constraints from ss-NMR data, several structure models of A β fibrils have been proposed.
Based on ss-NMR data, Tycko group proposed the first structure model of the A β 1–40 fibril in 2002. [ 30 ] Thereafter, structure models for A β 1–42 [ 31 ] and A β 1–40 [ 32 ] fibrils were reported by the Riek and Tycko groups, respectively. Since then, fibril structures of other amyloidogenic proteins or protein segments have been proposed, such as the structure models of hIAPP, [ 33 ] Tau proteins, [ 34 ] alpha-synuclein [ 35 ] fibrils. All of these fibrillar structures share a common cross- β structure feature in spite of their low sequence similarity. A β fibrils are mostly organized in parallel β -sheets, but antiparallel ones also exist. [ 36 ] Under different fibrillation conditions, A β 1–40/1–42 monomers can assemble into polymorphic fibrils with different symmetries. [ 37 ]
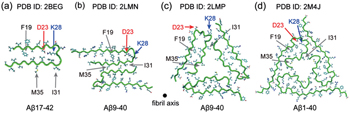
Four extensively studied ss-NMR-derived A β fibrils are given in Fig.
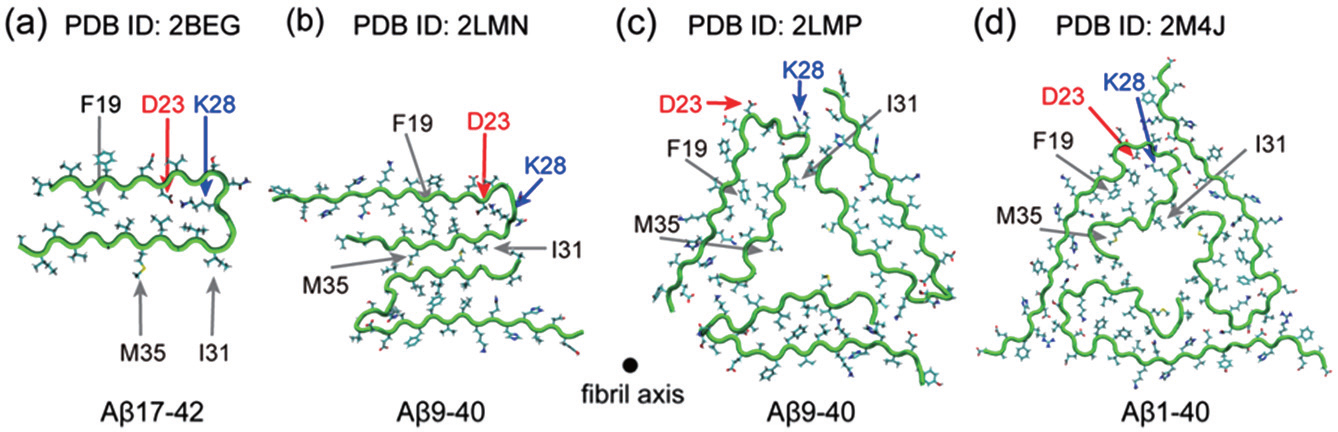 | Fig. 3. Four A β fibril structural models derived from ss-NMR. (a) A β 17–42 (PDB id: 2BEG), [ 31 ] (b) A β 9–40 with 2-fold symmetry (PDB id: 2LMN), [ 32 ] (c) A β 9–40 with 3-fold symmetry (PDB id: 2LMP), [ 37 ] (d) A β 1–40 fibril from the brain of AD patients, with 3-fold symmetry (PDB id: 2M4J). [ 38 ] |
Molecular simulations on multi-level models are widely used in various fields, including physics, chemistry, and biology. The Noble Prize in Chemistry in 2013 was awarded to three scientists who developed multi-scale models for molecular simulations. For biomolecular systems, molecular simulations are powerful tools to study amyloidogenic peptide aggregation on different levels, ranging from the most precise quantum calculation to coarse-grained (CG) models. Computational studies of peptide aggregation on the atomistic level provide a balance between accuracy and computational cost and become prolific tools. In 2011, Shaw et al . performed a series of MD simulations on Anton with the AMBER ff99SB-ILDN force field and proved that the current force field performs well in general. [ 39 ]
Various computational techniques have been employed to study amyloidogenic peptide aggregation (reviewed in a recent paper [ 40 ] ). The replica-exchange molecular dynamics (REMD) simulation method is particularly suited to sample the conformational space of short peptides The REMD method is an enhanced sampling protocol, in which several identical copies (replicas) of the system are simulated in parallel at different temperatures or at the same temperature but using different Hamiltonian and these replicas are periodically swapped with a probability given by the Metropolis criterion. This can avoid getting trapped in local-minimum free energy states. [ 41 ] The REMD method is useful for simulating complex problems such as protein folding and peptide aggregation. Since 2005, this method has been widely used in the study of amyloidogenic peptide aggregation. [ 42 – 44 ]
Due to its low computational cost, CG models have also been widely used in peptide aggregation, such as the OPEP [ 45 ] and PRIME models. [ 46 ] A detailed summary of the CG model used in peptide aggregation can be found in a recent review by Wu and Shea. [ 47 ] Here, we discuss recent progress of simulation studies using atom-level models by focusing on the oligomerization of A β peptides and its different fragments. Summaries of MD studies in this area in early years can be found in other reviews. [ 48 , 49 ]
Certain fragments of amyloidogenic peptides have similar properties as their full-length counterpart and can aggregate into β -sheet-rich fibrils in vivo and retain their toxicity. These fragments are used widely as model systems to study peptide aggregation. Here, we briefly review MD studies on the monomeric and oligomeric forms of different A β fragments.
As early as 2002, Ma and Nussinov examined the structural stability of various constructed fibril models of different A β fragments (including A β 16–22, A β 16–35, and A β 10–35) by MD simulations with the aim of characterizing the atomic structure of stable fibrils. Their MD simulations indicate that an antiparallel β -sheet orientation is the most stable for the A β 16–22 peptide, consistent with a solid-state ss-NMR-based fibril model of the A β 16–22 peptide. [ 50 ] A β 16–22 is a peptide that contains the central hydrophobic core (CHC) motif (LVFFA) of full-length A β . For A β 16–35 peptide, a bent double-layered hairpin-like parallel β -sheet structure was found to have higher structural stability. Residues D23 and K28 in the middle polar region of A β 16–35 form an intra-chain salt-bridge and stabilize the double-strand conformation. [ 51 ] The effects of mutation on the fibril structural stability of A β fragments were also investigated. By building various parallel/antiparallel arrangements of β -sheet oligomers, Ma et al. compared the stability of the A β 25–35 N27Q mutant with that of the wild type species [ 52 ] and found that a single mutation could reduce amyloid fibrillation propensity. MD simulations of the dissociation of A β 16–22 monomers from preformed fibrils [ 53 ] were also reported.
Numerous computational studies were initially focused on the conformational distribution of monomeric A β fragments. In 2006, Gnanakaran et al . explored the conformational space of the A β 16–22 peptide by performing REMD simulations, and found that the predominant secondary structure is the PPII rather than the β -strand (PPII content > 40%). [ 54 ] In the same year, using REMD simulations, Wei and Shea studied the free energy landscape of monomeric A β 25–35 in both pure water and HFIP/water cosolvent. In water, the A β 25–35 peptide mainly adopts collapsed coil conformations and to a lesser extent β -hairpins with a turn at G29–A30, while in the apolar organic HFIP solvent, this peptide preferentially populates a helical structure. [ 55 ]
The conformational distribution of various C-terminal fragments of A βx –42 ( x = 29–31, 39) were also studied by REMD simulations. These C-terminal A β fragments mostly adopt metastable β -hairpin conformations, except for A β 39–42, which has an extended β -strand structure. Interestingly, when residues 40 and 41 are truncated from the A β 30–42 fragment, the conformation shifts from β -hairpin to turn-coil state. [ 56 ] The folding energy landscape of the turn-region-containing peptide A β 21–30 was studied by Baumketner et al. using REMD simulations. [ 42 ] Salt bridges were found to be formed between residues E22 and K28, instead of between D23 and K28. Using the same method, they also studied the influence of the E22/D23 mutation on the monomeric structures of A β 21–30 and found that the E22 mutation does not alter the bend conformation while the D23 mutation converts the bend motif into a turn structure. [ 57 ] Similar results were also reported by Tarus et al. [ 58 ] Longer A β fragments, such as A β 10–35, [ 59 – 61 ] A β 12–28, [ 62 ] A β 12–36, [ 63 ] and A β 15–28, [ 64 ] have also been studied by MD simulations. Their relatively long aa sequences allow them to adopt multiple conformations, rather than having a single structure motif. [ 62 , 64 , 65 ]
A dimer is the smallest oligomer. Understanding the dimerization process of A β fragments provides structural insight into the early stage of amyloid fibrillation. In early years, due to the limitations of computational capability, dimeric systems became good model systems for understanding peptide aggregation. For example, in 2006, Gnanakaran et al . investigated the dimeric structures of A β 16–22 by conducting atomistic REMD simulations. They found that A β 16–22 dimers adopt various conformations, including parallel and antiparallel β -sheet structures. [ 54 ] The dimeric structures of A β 25–35 were also studied with REMD simulations. [ 66 , 67 ] Various structures coexist in a balance of disordered compact states and extended fibril-like β -sheet conformations including parallel and antiparallel β -sheets, β -hairpins, and U-shaped β -sheets. Moreover, the stable parallel and antiparallel bilayer β -sheet protofibrils identified by MD simulations are consistent with experimental data from H/D exchange NMR and atomic force microscope spectroscopy. [ 67 ] Energetic analysis reveals that the formation of compact conformations is driven by entropy, while the fibril-like state is stabilized by inter-/intrachain electrostatic interactions. [ 66 ] The dimerization of other A β fragments has also been studied by different simulation methods. [ 68 , 69 ]
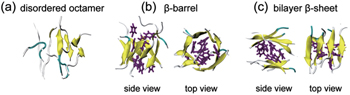
The increase of computational power in recent years allows researchers to investigate the conformational space of oligomers consisting of multiple peptide chains. For example, Li et al. explored the conformational distribution of A β 16–22 octamers by performing explicit-solvent REMD simulations in a neutral-pH condition in 2011. They found that A β 16–22 octamers adopt mostly disordered coil aggregates, and to a lesser extent, closed β -barrels and bilayer β -sheets. [ 70 ] Interestingly, one year later, the atomic view of cylindrical β -barrels formed by an 11-residue peptide was reported in an experimental study. [ 71 ] The octameric structures of A β 16–22 in an acidic pH condition were studied by Xie et al. using explicit-solvent REMD simulations. [ 72 ] Similar to the octameric conformations at neutral pH, [ 70 ] disordered aggregates and ordered octamers (including closed β -barrels and bilayer β -sheets) were observed (see Fig.
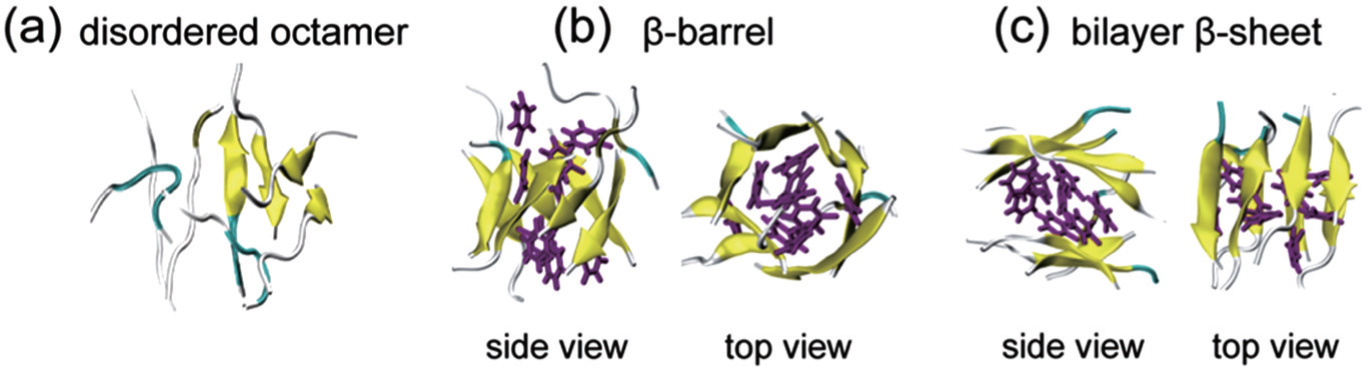 | Fig. 4. Representative structures of A β 16–22 octamers. (a) a disordered state, (b) a closed β -barrel structure in two different views, and (c) a bilayer β -sheet in two different views. The side chains of residues F19 and F20 are in purple stick representation. This figure is adapted from Ref. [ 72 ]. |
The influence of force fields on the structures of A β fragments has been investigated by several groups. For example, Wang et al . studied the conformations of monomeric A β 12–28 using two different models—OPLS–AA/TIP4P and GROMOS 43A1/SPC). [ 74 ] Structural analyses reveal that both models generate mostly random coil conformations with differences in the turn region of the β -hairpin (GROMOS 43A1: F19–E22; OPLS–AA: L17–F20). Thereafter, Nguyen et al . examined the structures of monomers, dimers, and trimers of A β 16–22 with AMBER99, GROMOS96, and OPLS force fields. [ 75 ] Their REMD simulation results show that AMBER99 favors a helical structure, GROMOS96 has a bias to antiparallel β -sheet conformations, and OPLS reveals diverse structures. The different conformational bias of these force fields reminds us to be cautious when choosing a force field.
Much effort has also been devoted to exploring the free energy landscape of the full-length A β peptide. For example, by performing implicit-solvent REMD simulations, Baumketner et al . found that A β 42 monomer adopts extended structures consisting mainly of loops and turns and to a lesser extent helices in the C-terminal region. [ 76 ] This work demonstrates the feasibility of the REMD method in sampling the conformational space of the monomeric state of a full-length A β peptide. Soon after, many researchers focused on the study of conformational and thermodynamic properties of monomeric A β using REMD simulations. [ 77 – 83 ] Besides A β 1–40/A β 1–42 peptides, [ 77 , 79 , 81 , 82 ] A β peptides containing 39 [ 78 ] and 43 [ 83 ] residues were also analyzed. Comparing the conformational ensembles and free energy landscapes of the A β monomer, the differences between these four sequences are summarized as follows.
MD and REMD simulations were also applied to compare various mutants (E22Q, [ 85 ] D23N, [ 86 ] D7H, [ 87 ] A2T/A2V [ 88 ] ) with wild type A β . Simulation results show that these mutations significantly alter the conformational landscape of the A β monomer. [ 85 ]
The REMD method was also used to study full-length A β oligomers. In 2010, Kim et al . simulated A β 1–40 monomers, dimers, and tetramers using implicit-solvent REMD simulations. Conformational analyses reveal sharp structural difference between monomers and oligomers. [ 89 ] Recently, the dimeric structures of full-length A β 1–40/A β 1–42 have been investigated using extensive explicit-solvent REMD simulations. [ 90 – 92 ] These studies report that the full-length A β dimer adopts various conformations with low α -helix and β -sheet content.
Knowing whether an A β fragment in its isolated form and this fragment within the full-length A β adopts similar conformations is important for understanding the role of the smaller fragment on the structure of full-length A β . A combined REMD and NMR study on A β 1–42 and A β 21–30 shows that A β 1–42 adopts various conformations spanning from collapsed structured states to highly extended conformations, while A β 21–30 has a relatively homogeneous ensemble of extended structures. This result indicates that A β 21–30 in isolation and the 21–30 region in full-length A β has different conformational space. [ 93 ]
During the aggregation process of A β peptides, when additional factors (such as nanoparticles, metal ions, [ 94 ] and membranes) are involved in this process, they can modulate A β fibrillation. The regulation of A β aggregation by metal ions and membranes has been discussed in a recent review. [ 95 ] Here, we discuss only the effect of carbon-based nanoparticles on A β aggregation.
Nanoparticles have been reported to be able to influence amyloid formation. [ 96 , 97 ] There are three different kinds of carbon nanoparticles: carbon nanotubes, fullerenes, and graphites. Kim et al . found that fullerenes could inhibit the aggregation of A β 11–25 and A β 1–40. [ 98 ] Modified fullerenes were also able to prevent the aggregation of A β 25–35 peptide. [ 99 , 100 ] Linse et al . reported that carbon nanotubes could enhance the fibril formation of the globular protein β 2-microglobulin. [ 27 ] Graphites were shown to promote the formation of A β fibrils. [ 101 – 103 ]
In spite of a series of experimental studies, the molecular mechanism of carbon-nanoparticle-modulated aggregation of A β peptides is unclear. Here, we review recent atomistic MD studies on this topic. The atomistic MD study by Fu et al . in 2009, showed that a single-walled carbon nanotube (CNT) could induce two preformed bilayer β -sheets with mixed antiparallel–parallel strands to assemble into a closed β -barrel wrapping the CNT. By analyzing the A β 16–22–CNT interactions, the authors suggested that the β -barrel formation on SWNT surface results from the interplay of dehydration, peptide–CNT, and peptide–peptide interactions. [ 104 ] Two years later, Li et al . studied the interaction of another A β fragment–A β 16–22 with CNT by REMD simulations in explicit solvent. [ 70 ] Their simulations showed that CNTs not only prevented the formation of β -sheet-rich oligomers, but also destabilized prefibrillar β -sheets via hydrophobic and π – π stacking interactions. Following the above-mentioned two simulation studies, Xie et al . examined the influence of the hydroxylated CNT (CNT-OH) on A β 16–22 aggregation using REMD simulations. [ 105 ] Similar to the pristine CNTs, the hydroxylated ones could also inhibit the formation of ordered β -sheet-rich structures. Besides the hydrophobic and aromatic stacking interactions, strong electrostatic interactions between the hydroxyl groups of CNTs and the positively charged residue K16 of A β 16–22 also played an important role in β -sheet inhibition. Atomic force microscopy and thioflavin T fluorescence experiments by the same authors provide direct evidence of the inhibitory effect of both pristine and hydroxylated CNTs on A β 16–22 fibrillization.
The absorption of helical A β 1–42 monomer on CNT surface was studied by Jana et al . using MD simulations and the simulations showed that helical A β 1–42 could be rapidly absorbed on the surface of the nanotube. The CHC region of A β was identified as the most favorable interaction site. [ 106 ] In addition, the importance of F19 in the interaction of A β 1–42 with CNT were also examined by the same group with MD simulations of A β 1–42 mutants. [ 107 ]
With the advent of MD simulation studies on A β –CNT system, the influence of fullerenes on the structural stability of A β protofibrils was also explored by MD simulations. In 2012, Andujar et al . studied the interaction between fullerene and A β fibrillar structure by the docking method and MD simulations. [ 108 ] They found that the binding of C60 to the CHC site caused significant structural alterations of the A β protofibrils. Using similar methods, Huy et al . studied the binding affinity of A β fibril with different sizes of fullerenes ranging from C20 to C84. [ 109 ] Compared to other fullerenes, the fullerene C60 is more energetically favorable. In addition, fullerenes have higher binding affinity with A β 9–40 than with A β 17–42.
The disruption effect of a soluble fullerene derivative on the A β 1–42 fibril was investigated by Zhou et al . using MD simulations. [ 110 ] Three dominant binding sites were identified: the CHC site ( 17 LVFFA 21 ), the turn site ( 27 NKGAI 31 ), and the C-terminal β -sheet site ( 31 IIGLMVGGVVI 41 ). The CHC site is consistent with the one reported previously in the docking study by Andujar et al. [ 108 ] The other two binding sites, the turn site, and the C-terminal β -sheet site, are newly identified binding sites. [ 110 ] Both the aromatic stacking interaction in the CHC site and the curvature matching between fullerene and A β 1–42 protofibrils played a crucial role for fullerene binding. The binding of fullerene to the turn region disrupted the D23–K28 salt-bridge that is important for the A β fibrillation. These simulation results provide an atomic-level explanation for experimentally-observed inhibitory effect of fullerene on A β fibrillation. [ 98 ]
Conventional MD simulations can be easily trapped in the local free-energy-minimum state. To enhance the conformational sampling, Xie et al. , by using REMD simulations, studied the effect of C60 on A β 16–22 octamerization. [ 111 ] Their simulations demonstrated that fullerenes could significantly weaken the inter-peptide interaction and reduce β -sheet propensity, thus leading to disordered oligomers. Interestingly, they found that one C180 molecule displayed stronger inhibitory effect on the β -sheet formation than three C60 molecules (3C60 for short) although they contain the same number of carbon atoms. Fullerene–peptide interaction analyses revealed that the stronger inhibitory effect of C180 on β -sheet formation results from strong hydrophobic and aromatic-stacking interactions between fullerene hexagonal rings and the Phe rings (C180 contains many more hexagonal rings than 3C60).
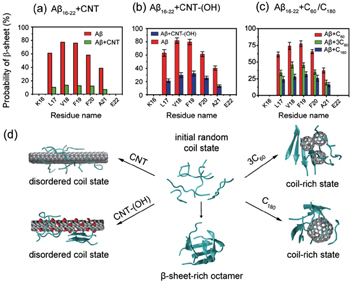
The influence of carbon nanoparticles (including CNT, CNT-OH, C60, 3C60, and C180) on A β 16–22 oligomerization is summarized in Fig.
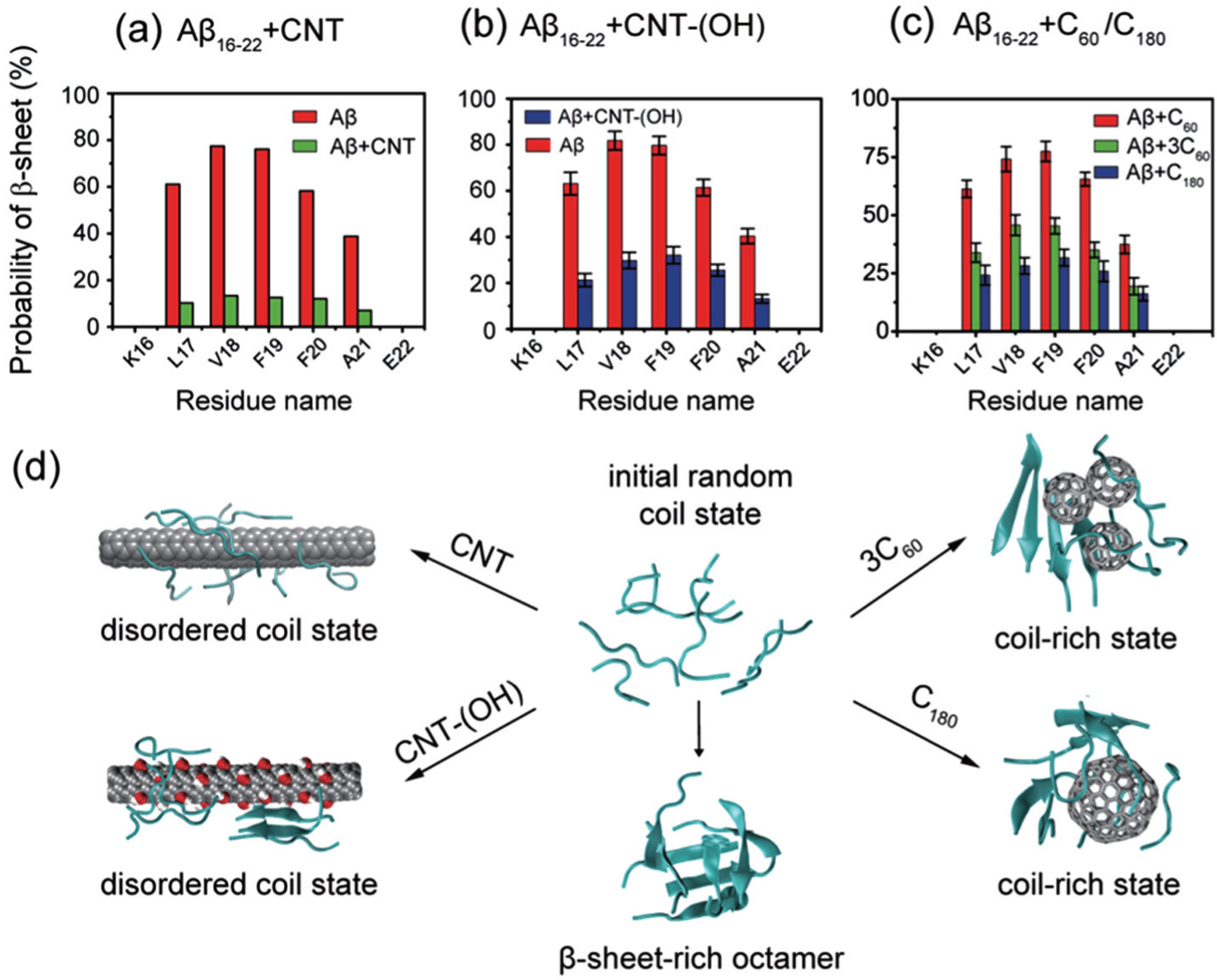 | Fig. 5. Effects of different carbon nanoparticles on the oligomerization of A β 16–22 peptide. The β -sheet propensity of A β 16–22 octamers is reduced by (a) pristine CNT, (b) CNT-(OH), and (c) three C60 and one C180. Pristine CNTs display the strongest inhibitory effect. (d) Representative conformations of A β 16–22 octamers in the absence and presence of four different carbon nanoparticles. |
Atomistic MD simulation studies of A β aggregation have achieved a great success in various aspects from oligomeric structures to aggregation mechanism in recent years. The REMD method is one of the most suitable strategies to study aggregation of unstructured amyloidogenic peptides. Conformational sampling by REMD was proved to be efficient for short A β fragments. Simulation results demonstrate quantitative comparability with experimental results. However, REMD simulations can only provide structural and thermodynamic properties of small oligomers. The kinetics of peptide aggregation is mostly unknown at the atomic level. It is still a challenge for atomistic simulations to uncover the oligomerization process of full-length A β . From 2006 to 2015, the simulated system was extended from oligomers of peptide fragment (< 20 aa) to dimers of longer A β fragments and full-length A β . In the next decade, computational capabilities might enable us to carry out REMD simulations on the oligomerization of multiple full-length A β or other peptides. On the one hand, developments in computing ability would provide much information on the early stage of peptide aggregation. On the other hand, more structural information relies on the advances in experimental methods for structural characterization of oligomers. With the rapid development of computing power, improvement of sampling methods, protein force fields, and experimental methods, the processes of A β oligomerization, fibril elongation, [ 112 – 115 ] cross-seeding, [ 116 , 117 ] and amyloid inhibition will be uncovered in the near future.
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 | |
| 23 | |
| 24 | |
| 25 | |
| 26 | |
| 27 | |
| 28 | |
| 29 | |
| 30 | |
| 31 | |
| 32 | |
| 33 | |
| 34 | |
| 35 | |
| 36 | |
| 37 | |
| 38 | |
| 39 | |
| 40 | |
| 41 | |
| 42 | |
| 43 | |
| 44 | |
| 45 | |
| 46 | |
| 47 | |
| 48 | |
| 49 | |
| 50 | |
| 51 | |
| 52 | |
| 53 | |
| 54 | |
| 55 | |
| 56 | |
| 57 | |
| 58 | |
| 59 | |
| 60 | |
| 61 | |
| 62 | |
| 63 | |
| 64 | |
| 65 | |
| 66 | |
| 67 | |
| 68 | |
| 69 | |
| 70 | |
| 71 | |
| 72 | |
| 73 | |
| 74 | |
| 75 | |
| 76 | |
| 77 | |
| 78 | |
| 79 | |
| 80 | |
| 81 | |
| 82 | |
| 83 | |
| 84 | |
| 85 | |
| 86 | |
| 87 | |
| 88 | |
| 89 | |
| 90 | |
| 91 | |
| 92 | |
| 93 | |
| 94 | |
| 95 | |
| 96 | |
| 97 | |
| 98 | |
| 99 | |
| 100 | |
| 101 | |
| 102 | |
| 103 | |
| 104 | |
| 105 | |
| 106 | |
| 107 | |
| 108 | |
| 109 | |
| 110 | |
| 111 | |
| 112 | |
| 113 | |
| 114 | |
| 115 | |
| 116 | |
| 117 |