{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Strategies to curb structural changes of lithium/transition metal oxide cathode materials & the changes’ effects on thermal & cycling stability
[Yu Xiqian , Hu Enyuan , Bak Seongmin , Zhou Yong-Ning , Yang Xiao-Qing †,  ]
]
]
|
|


† Corresponding author. E-mail:
Project supported by the U.S. Department of Energy, the Assistant Secretary for Energy Efficiency and Renewable Energy, Office of Vehicle Technologies (Grant No. DE-SC0012704).
Structural transformation behaviors of several typical oxide cathode materials during a heating process are reviewed in detail to provide in-depth understanding of the key factors governing the thermal stability of these materials. We also discuss applying the information about heat induced structural evolution in the study of electrochemically induced structural changes. All these discussions are expected to provide valuable insights for designing oxide cathode materials with significantly improved structural stability for safe, long-life lithium ion batteries, as the safety of lithium-ion batteries is a critical issue; it is widely accepted that the thermal instability of the cathodes is one of the most critical factors in thermal runaway and related safety problems.
Lithium-ion batteries (LiBs) have been leading candidates for vehicle applications due to their high energy density and high power capability. [ 1 – 4 ] However, LiBs’ safety issues have to be soundly addressed before large scale application. [ 5 – 7 ] When an LiB experiences certain abusive situations, for example, shorting, the temperature of the battery can easily rise to the threshold of so-called “thermal runaway,” in which the temperature rises very rapidly and is out of control. [ 6 ] A series of chemical reactions are triggered during such a process, and a considerable amount of heat can be released, possibly leading to fire or explosion. Each component in the battery, including anode, [ 8 – 10 ] separator, [ 11 – 13 ] electrolyte, [ 14 – 17 ] and cathode, [ 18 – 23 ] has its role to play in this process, and the role of the cathode has been proved to be particularly important. Only oxide systems as cathode materials are discussed in the present review. Readers interested in thermal stability of polyanion systems are encouraged to read references in the literature. [ 24 – 26 ]
Dahn et al. investigated the thermal stability of charged cathodes, including Li x NiO 2 , Li x CoO 2 , and λ -MnO 2 (fully charged state of LiMn 2 O 4 ), and found that they all release oxygen upon heating. [ 22 ] Arai et al. conducted DSC measurements for charged Li x NiO 2 , ethylene carbonate (EC, a major component of the electrolyte), and the combination of these two, [ 23 ] finding that heating the mixture of charged Li x NiO 2 and EC is much more exothermic than heating either of them individually. This suggests that oxygen-release from a cathode is a key factor contributing to heat generation of the whole reaction. More advanced cathode materials normally contain multiple metal elements. Thermogravimetric and calorimetric studies of charged Li x Ni 0.8 Co 0.15 Al 0.05 O 2 and charged Li x Ni 1/3 Co 1/3 Mn 1/3 O 2 reveal that the former releases a larger amount of oxygen at a lower temperature than the latter. [ 27 ] This correlates well with the fact that the former has much poorer safety characteristics in terms of a much greater amount of heat generated than the latter. [ 17 ] Such a relationship between oxygen-release and safety characteristics is further confirmed in conditions closer to real battery operation by testing full-battery cells which include anode, separator, electrolyte, and cathode. [ 28 ]
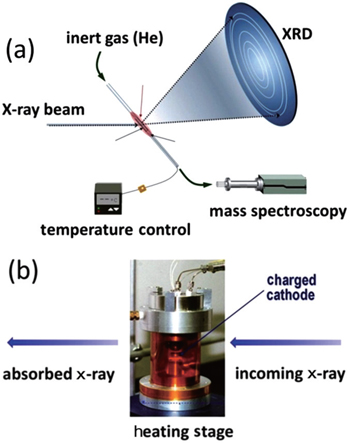
When a charged cathode is heated, it tends to release oxygen. This is considerably detrimental because the released oxygen can react with the electrolyte in a highly exothermic way, significantly accelerating the elevation of temperature, and initiating further disastrous reactions. Therefore, designing cathode materials with suppressed oxygen-release and/or increased onset temperature for oxygen-release would be an effective approach to such safety issues. While thermogravimetric, [ 9 ] calorimetric, [ 23 ] and computational studies [ 29 ] have provided useful information like heat generation rate, chemical reaction kinetics, and so on; it is desirable to understand the roles of crystal and electronic structural changes in the oxygen-release process, especially through in situ techniques. Hence, our group designed in situ x-ray diffraction (XRD) and in situ x-ray absorption (XAS) techniques for studying the thermal stability of charged cathode materials. Here, we start with the experimental setup, then review results from our studies, [ 30 – 35 ] and finally discuss the implications of these results for designing a stable cathode.
A technique combining in situ x-ray diffraction with mass spectroscopy (MS) during heating was developed at the beamline 7B of the National Synchrotron Light Source (NSLS) facility by our group. [ 32 ] This technique enables us to monitor crystal structure changes and oxygen-release simultaneously. In an in situ XRD-MS experiment illustrated in Fig.
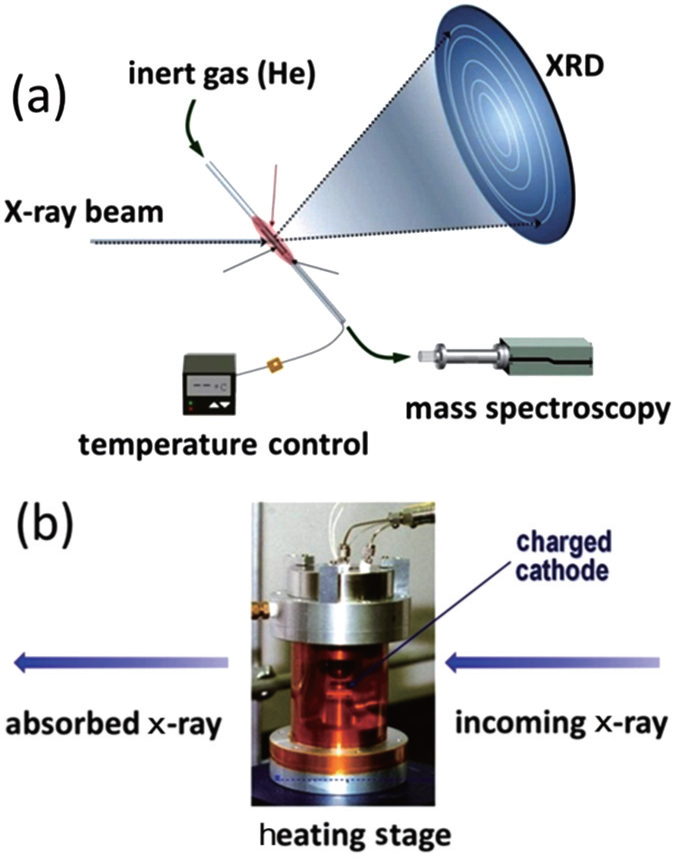 | Fig. 1. (a) In situ XRD-MS and (b) in situ XAS experimental setup. The figure is adapted from Ref. [ 32 ]. |
Different contents and concentration ratios of metal elements in the cathode materials result in different thermal stability of the cathodes. This suggests that each element can play a unique role in stabilizing or destabilizing the structure. Commonly used metal elements for lithium-ion battery cathodes are mainly 3d transition metal elements, which are very close to each other in the periodic table, making it difficult to distinguish them by XRD alone. Therefore, x-ray absorption is a very valuable tool in differentiating them. An XAS spectrum includes both the x-ray absorption near edge structure (XANES) and the extended x-ray absorption fine structure (EXAFS). These parts can provide information about the oxidation state, electronic structure, and local environment in an elemental selective way. An in situ XAS technique (shown in Fig.
Cubic-close-packed oxygen anions are layered, with lithium and transition metal occupying the octahedral sites in alternating layers. Among the layer-structured cathode materials, LiNi 0.8 Co 0.15 Al 0.05 O 2 and LiNi 1/3 Co 1/3 Mn 1/3 O 2 are important due to their useful electrochemical performance. However, despite the similarity of their crystal structures, their thermal stabilities differ quite a bit, suggesting the important role of the elemental content. Our group studied these two materials systematically, and some of the major results are reviewed here.
Li x Ni 0.8 Co 0.15 Al 0.05 O 2 is highly favored for high energy applications. However, as suggested by its high nickel content, this material features poor thermal stability. The previous studies showed that this material releases oxygen at temperatures as low as around 200 °C, lower than the oxygen-release onset of Li x CoO 2 and λ -Mn 2 O 4 (charged LiMn 2 O 4 ). [ 22 ] Structural investigations indicate that phase changes at high temperatures follow the “layered to spinel, and then to rock salt” path. [ 20 , 21 ] What is fundamentally important but has not been studied in detail is the relationship between the oxygen-release phenomena at the macroscopic level and the crystal structure changes at the atomic level, as well as the contribution of each individual element to these changes. In this review, through the results of in situ XRD-MS and in situ XAS studies, the correlations among crystal structure changes, transition metal migration, and oxygen-release will be thoroughly discussed, and more detailed information can be obtained from our previous publications. [ 31 ]
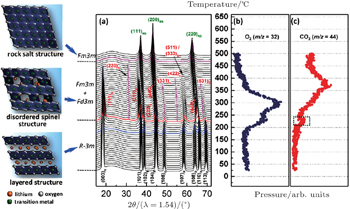
In situ XRD-MS results for the overcharged Li x Ni 0.8 Co 0.15 Al 0.05 O 2 ( x = 0.33) are shown in Fig.
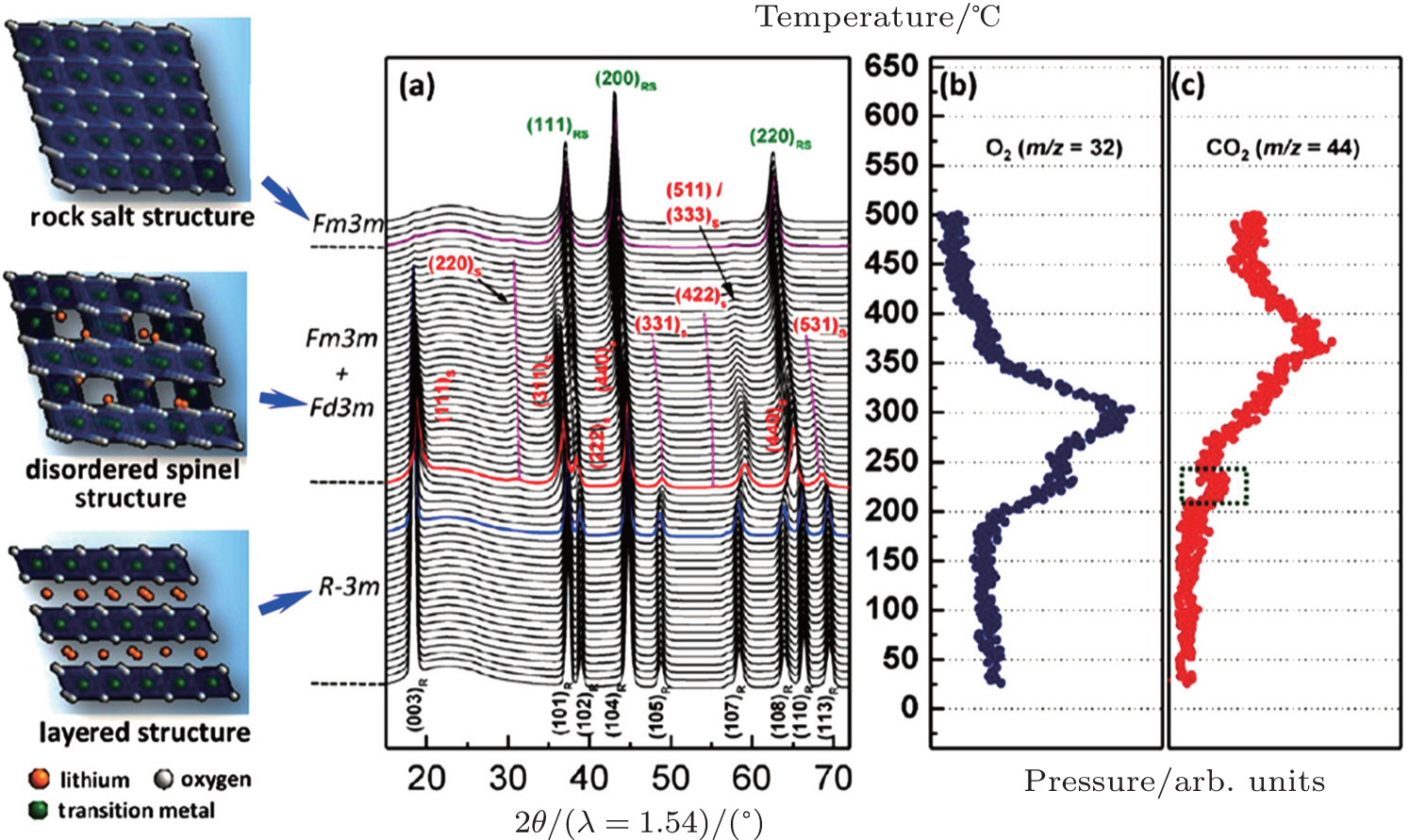 | Fig. 2. (a) TR-XRD patterns and simultaneously measured mass spectra for (b) O 2 and (c) CO 2 , released from Li 0.33 Ni 0.8 Co 0.15 Al 0.05 O 2 during heating to 500 °C. The formation of CO 2 is associated with the oxidation of carbon (from either the PVDF binder or the conducting carbon in the charged electrode) by the released oxygen. [ 31 ] |
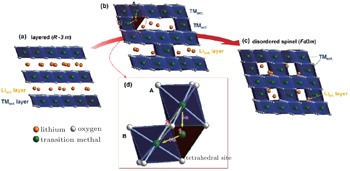
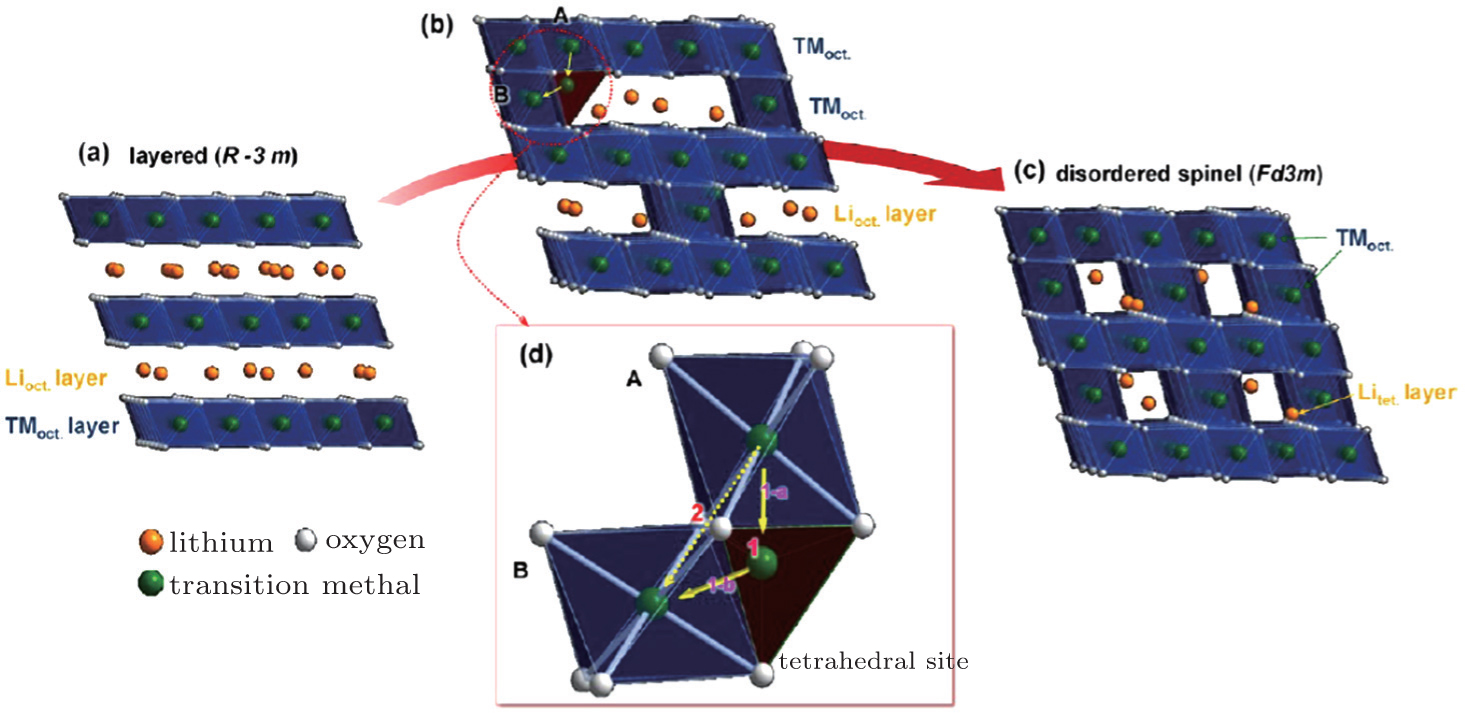 | Fig. 3. Phase transition of Li x Ni 0.8 Co 0.15 Al 0.05 O 2 charged cathode during heating: (a) layered structure, (b) cation migration during the phase transition from layered structure to disordered-spinel structure, (c) disordered spinel structure, and (d) cation migration path from octahedral A to octahedral B. Direct migration (path 2) between octahedral sites is energetically unfavorable, so transition metal ions prefer to travel through a neighboring tetrahedral site to the octahedral site (path 1). [ 31 ] |
LiNi 1/3 Co 1/3 Mn 1/3 O 2 was proposed by Ohzuku et al. in 2001, [ 36 , 37 ] showing promising electrochemical performance and interesting structural features. This material demonstrated good capacity, good rate capability (200 mA·h·g −1 at 18.3 mA·h·g −1 and 150 mA·h·g −1 at 1600 mA·h·g −1 ), [ 38 ] and good thermal stability. [ 19 ] Since then, various derivatives of this material have been explored, [ 39 – 41 ] mainly by varying the nickel, cobalt, and manganese contents in a random or specified way (e.g., LiNi 0.5− x Co 2 x Mn 0.5− x O 2 ).
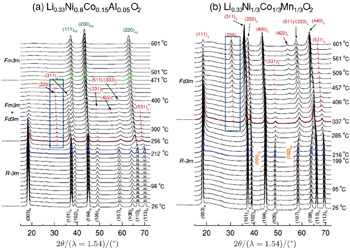
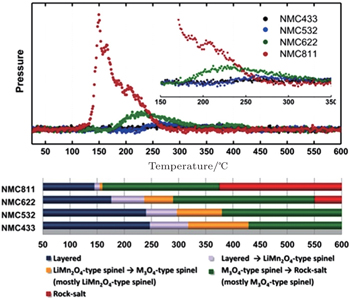
In our previous comparative studies, [ 32 ] charged Li x Ni 1/3 Co 1/3 Mn 1/3 O 2 ( x = 0.33, overcharged NCM) was shown to be much more stable than charged Li x Ni 0.8 Co 0.15 Al 0.05 O 2 ( x = 0.33, overcharged NCA). At elevated temperature, the NCM releases considerably less oxygen than the NCA. Correspondingly, the NCM involves only the layered to spinel phase transition, with the spinel phase well preserved up to 500 °C. In contrast, the NCA involves both the layered to spinel and the spinel to rock-salt transitions, with the second step releasing a large amount of oxygen. Such differences in structural changes can be clearly seen in Fig.
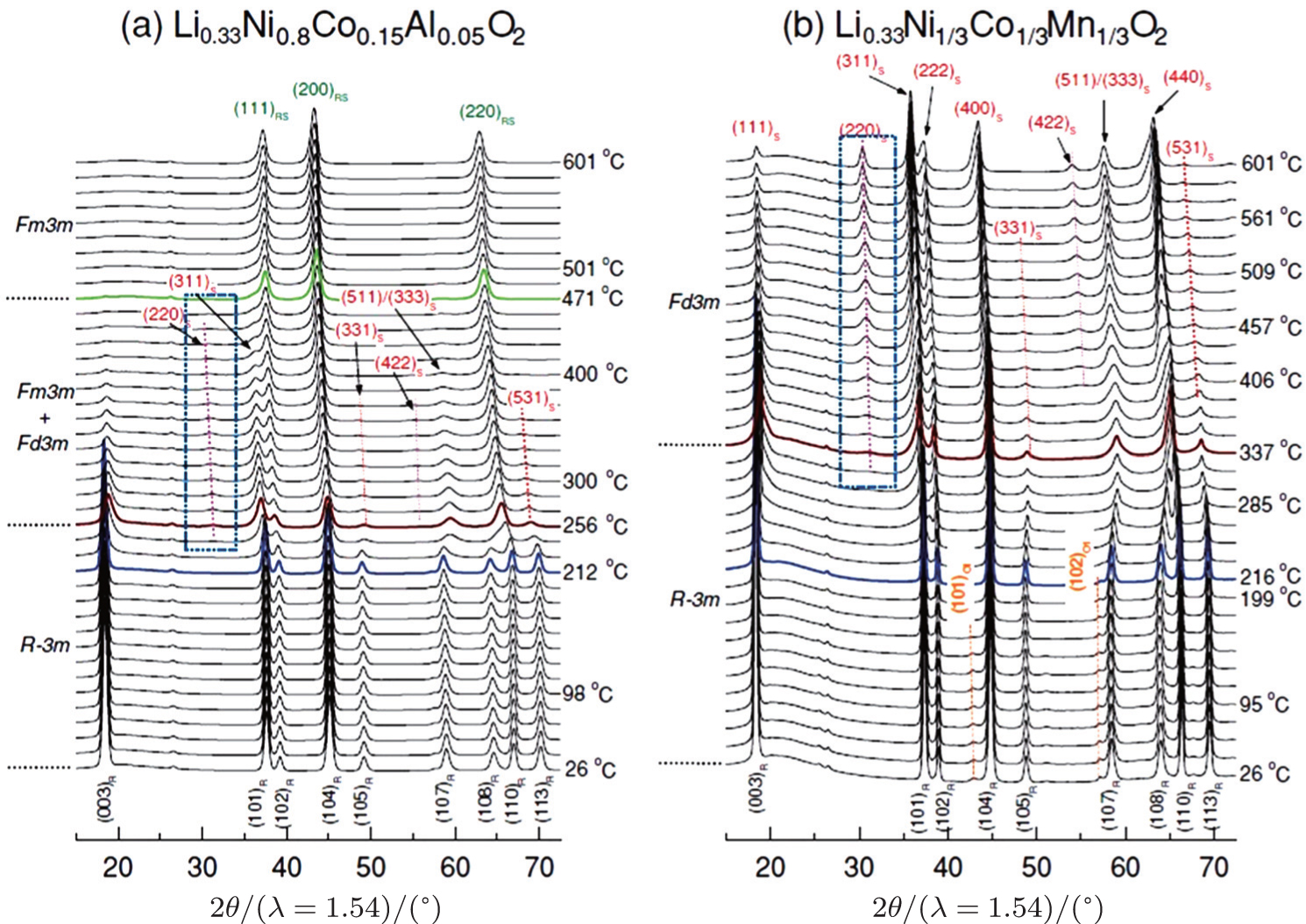 | Fig. 4. Time-resolved (TR) XRD patterns of overcharged (a) Li 0.33 Ni 0.8 Co 0.15 Al 0.05 O 2 and (b) Li 0.33 Ni 1/3 Co 1/3 Mn 1/3 O 2 during heating to 600 °C. The overcharged cathode samples sealed in quartz capillaries were heated from 25 °C to 600 °C for 4 h during the TR-XRD measurement (heating rate = 2.4 °C·min −1 ). The subscripts R, S, and RS denote rhombohedral, spinel, and rock-salt structures, respectively. The subscript O1 represents CdI 2 -type M O 2 ( M = Ni, Co Mn) structure. [ 32 ] |
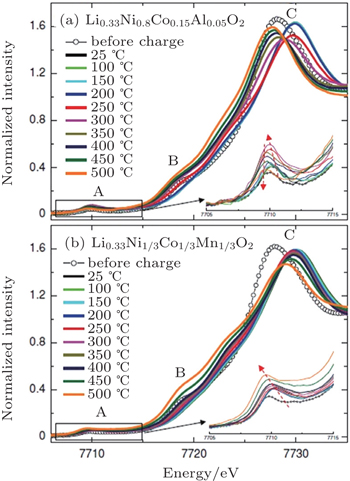
In our in situ XAS experiment, cobalt was identified to be the responsible element for the formation of the Co 3 O 4 -type spinel through migration to the tetrahedral sites for both Li 0.33 Ni 0.8 Co 0.15 Al 0.05 O 2 and Li 0.33 Ni 1/3 Co 1/3 Mn 1/3 O 2 . However, in the NCM case, cobalt was observed to stay at a tetrahedral site upon further heating, stabilizing the structure as Co 3 O 4 -type spinel up to 500 °C. This differs significantly from the behavior of cobalt in Li 0.33 Ni 0.8 Co 0.15 Al 0.05 O 2 , where cobalt ions migrate back to octahedral sites after spending a brief time at tetrahedral sites. Presumably due to the dilute concentration of cobalt, the Co 3 O 4 -type spinel structure in the Li 0.33 Ni 0.8 Co 0.15 Al 0.05 O 2 ’s case is relatively transient. Shortly after its formation, the rock-salt phase appears, initiating the step that involves significant oxygen release. The significant contrast in cobalt migration behavior is clearly seen in Fig.
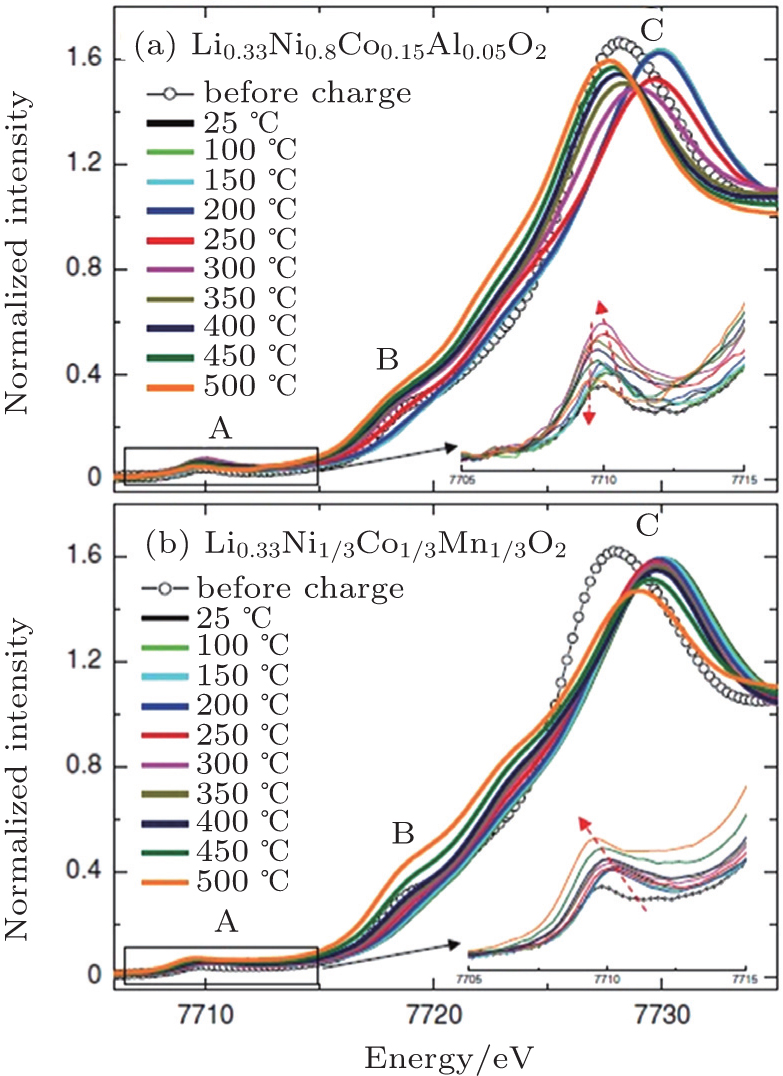 | Fig. 5. Cobalt K -edge XANES spectra of overcharged (a) Li 0.33 Ni 0.8 Co 0.15 Al 0.05 O 2 and (b) Li 0.33 Ni 1/3 Co 1/3 Mn 1/3 O 2 electrodes during heating up to 500 °C. Insets show the detailed feature of pre-edge region A. [ 32 ] |
This was further confirmed in studies of a series of charged LiNi x Co y Mn z O 2 samples with various nickel, cobalt, and manganese contents, [ 33 ] showing that higher cobalt content can suppress the oxygen-release more effectively, as seen in Fig.
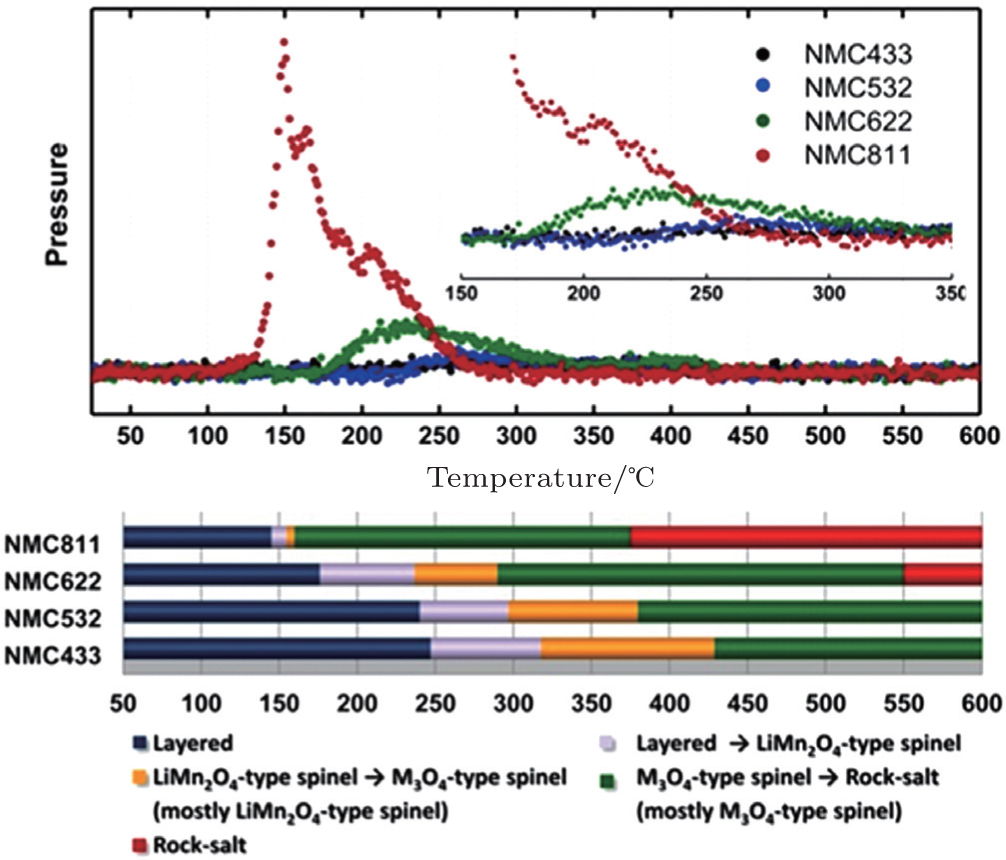 | Fig. 6. Mass spectroscopy profiles for oxygen (O 2 , m / z = 32), collected simultaneously during measurement of TR-XRD, and the corresponding temperature regions of the phase transitions for NCM samples (lower panel). [ 33 ] |
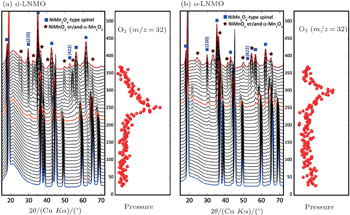
High voltage spinel LiNi 0.5 Mn 1.5 O 4 has attracted lots of attention in the past decade due to its high operating voltage (around 4.7 V), which translates into high energy density. [ 42 ] This material can either form the disordered phase with space group 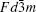
The high voltage spinel can be viewed as a derivative of the conventional spinel LiMn 2 O 4 with a quarter of the manganese replaced by nickel. After such substitution, the redox couple Mn 3+ /Mn 4+ in LiMn 2 O 4 , which is around 4.0 V, is replaced by the Ni 2+ /Ni 3+ and Ni 3+ /Ni 4+ redox couples in LiNi 0.5 Mn 1.5 O 4 , which are around 4.7 V. This is beneficial for high energy density. However, thermal stability of the cathode material seriously deteriorates after nickel substitution. For fully charged LiMn 2 O 4 , which is also referred to as λ -MnO 2 , no oxygen-release is observed up to temperatures around 400 °C. [ 22 , 52 ] However, charged LiNi 0.5 Mn 1.5 O 4 releases oxygen below 250 °C, with the ordered phase having slightly better thermal stability than the disordered one, as can be seen from Fig.
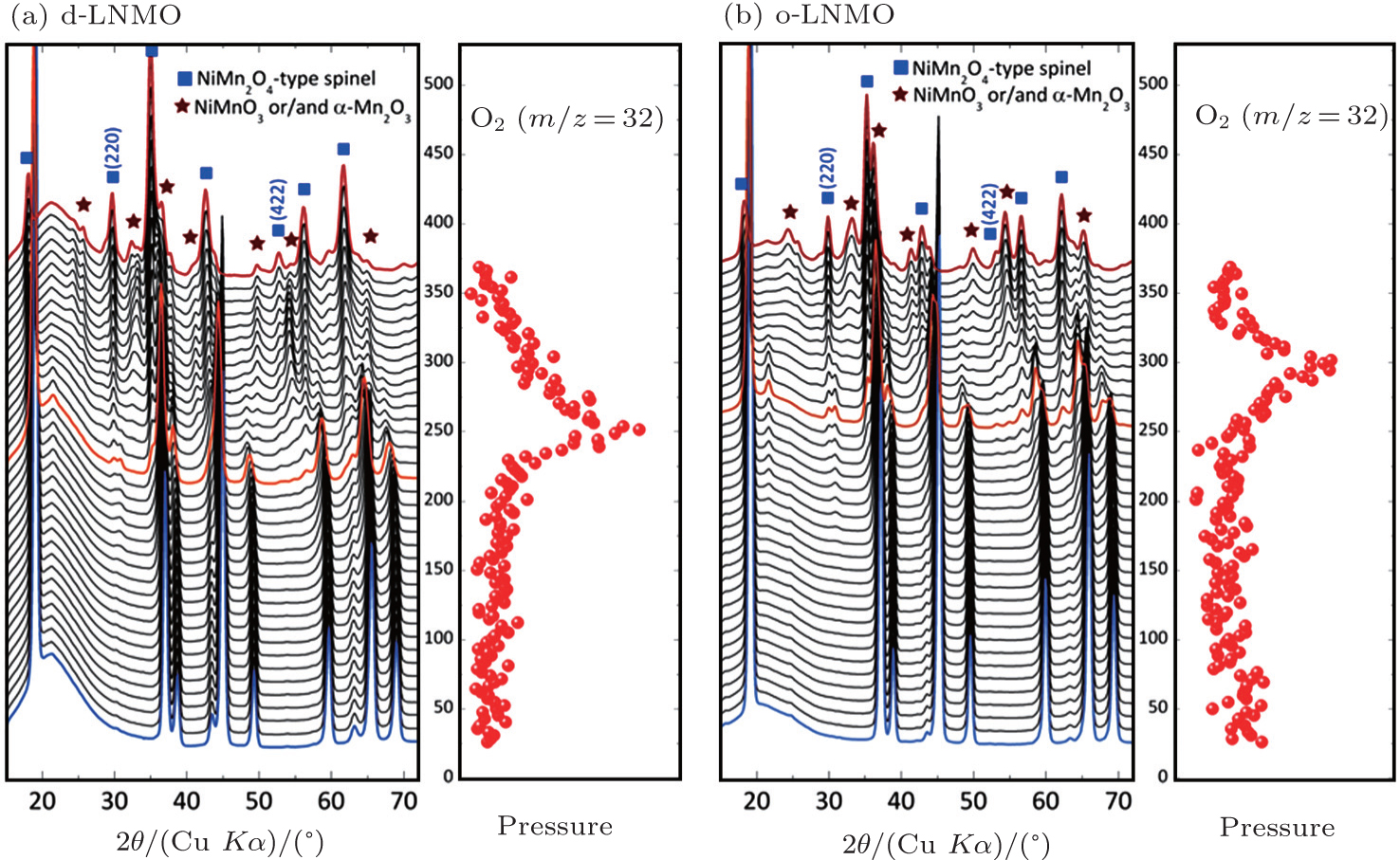 | Fig. 7. In situ XRD patterns combined with simultaneously measured mass spectroscopy data that trace the release of gaseous oxygen of (a) disordered charged LiNi 0.5 Mn 1.5 O 4 and (b) ordered charged LiNi 0.5 Mn 1.5 O 4 during heating to 375 °C. Left: in situ XRD patterns; right: profile of oxygen release. [ 34 ] |
This difference might be caused by the extra stability arising from cation ordering. In the P 4 3 32 phase, cations are arranged in an ordered way, releasing the strain and leading to a lower energy state. Such ordering is preserved in the charged sample and is believed to contribute to better thermal stability. In both cases, oxygen-release is accompanied by decomposition of the original crystal structure. The spinel framework is destroyed, decomposing into a mixture of NiMnO 3 , α -Mn 2 O 3 , and NiMn 2 O 4 . The former two have completely different cation arrangements from the original spinel framework. The latter one, NiMn 2 O 4 , has the same spinel framework as delithiated LiNi 0.5 Mn 1.5 O 4 , providing the possibility that only transition metal cation migration is required to change Li x Ni 0.5 Mn 1.5 O 4 ( x = 0) to NiMn 2 O 4 . In situ XAS studies reveal that nickel is quickly reduced, like in the Li x NiO 2 ’s case, and remains in the octahedral environment. Manganese, in contrast, migrates to the tetrahedral sites upon oxygen release, yielding the transition metal-in-tetrahedral site feature in NiMn 2 O 4 . Note that manganese has to be reduced from the original tetravalent state to a state close to the divalent state in order to enable such migration. Considering the large amount of manganese present in the sample, this implies that lots of reduction has to occur. In other words, considerable oxygen loss is inevitable in the phase transformation process.
From the above examples, the oxygen-release at high temperatures is an inevitable event for charged oxide cathode materials. Such inevitability can be understood from a thermodynamic point of view. [ 29 ] During the temperature elevation, the total amount of metal cations, including lithium and transition metals, is fixed but the amount of oxygen can vary. This implies that the phase diagram of multi-metal oxides with a fixed metal-to-metal ratio can provide the thermodynamic roadmap for the phase transition that occurs during temperature increase. (For instance, phase transitions of fully delithiated LiNi 0.5 Mn 1.5 O 4 at high temperature can be well understood by referring to the phase diagram of Ni/Mn oxide with a nickel-to-manganese ratio of 1:3.) Since, in these phase diagrams, the high temperature phase is normally low in oxygen compared to the low temperature phase, it is not surprising that oxygen-release can hardly be avoided for charged oxide cathode materials. In addition, the high oxygen partial pressures of oxides with highly oxidized cations such as Ni 4+ and Co 4+ imply that the cations all have a strong tendency to reduce. [ 53 ] Therefore, both the phase diagram and the high oxidation state of transition metals in the charged cathode samples can explain the thermodynamic origins of the oxygen-release.
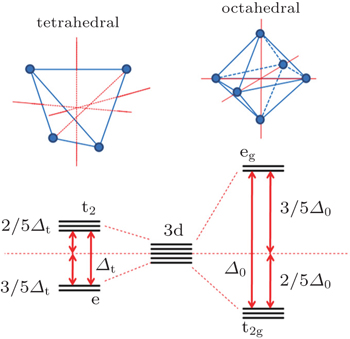
Since both structural change and transition metal reduction (and therefore oxygen-release) are thermodynamically driven, strategies for suppressing the oxygen-release leverage kinetic factors. In the charged oxide cathode materials that consist of Ni, Co, and Mn, the ions of Ni, Co, and Mn are oxidized close to Ni 4+ , Co x + (3.5 < x < 4), and Mn 4+ . [ 54 ] Among these, Ni 4+ is the easiest ion to reduce (to Ni 2+ ), while Mn 4+ the most difficult to reduced (to Mn 2+ ). Therefore, the reduction of the Ni 4+ ions takes place at the early stage of the heating process and generates a relatively large amount of oxygen gas. This is a critical step that governs the overall thermal instability of the material. To complete the structural transformation (such as layered to spinel and spinel to rock-salt) associated with the reduction of Ni 4+ , rearrangement of the Ni ions and other transition metal ions is necessary. This process requires migration of the transition metal ions from their original octahedral sites into either octahedral sites in neighboring layers or tetrahedral sites. [ 55 ] Direct migration between octahedral sites is energetically unfavorable because the transition metal ions face strong columbic repulsion on the travel path. They prefer to travel through the nearest tetrahedral sites as shown in Fig. 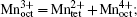
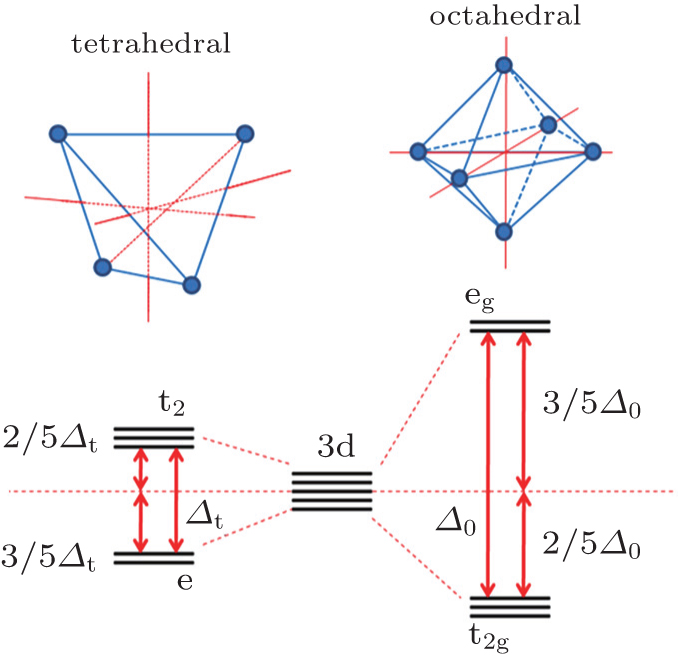 | Fig. 8. Crystal field splitting of the transition metal 3d orbital in tetrahedral and octahedral environments. The tetrahedral crystal field splitting energy Δ t is smaller than the octahedral crystal field splitting energy Δ 0 and yields the relationship Δ t = 4/9 Δ 0 . The site preference of the transition metal ion can be predicted by crystal field stabilization energy (CFSE) calculated based on its electron configuration. |
In summary, reduction of the transition metal ions, accompanied by oxygen-release and structural transformation, is unavoidable during the heating process in most cases. Since the structural transformation requires migration of the transition metal ions through tetrahedral sites, cations that need no or less reduction to become mobile and prefer to occupy tetrahedral sites favor overall thermal stability of the cathode materials. Such cations stabilize the structure at a certain phase at an early stage of the heating process, delaying further structural transformation to higher temperatures. Less oxygen gas will be generated, and oxygen-release will be spread across a wider temperature range. For example, some fixed valence cations (Mg 2+ , Al 3+ , Zn 2+ , etc.), migrate easily to tetrahedral sites during the heating process and have been proven to improve thermal stability of transition metal oxide cathode materials. However, these cations are electrochemically inactive, so a considerable amount of substitution will come at the expense of the capacity of the materials. Substitution of specific electrochemically active elements, such as Co and Fe, could be a better choice to enhance the thermal stability of oxide materials. This has been demonstrated in the above discussion of the NCA and NCM series materials and will be further studied in Fe-substituted high voltage spinel in our future work. [ 59 ]
The time-resolved XRD and XAS studies reveal that the structure and chemical composition of the materials play important roles in determining their structural stability during heating. These XRD and XAS data were collected by averaging a sample area on mm scale, therefore only reflecting the average structural changes. In fact, the structural transformation during the heating process involves nucleation and propagation of new structure, which takes place at the atomic scale. Exploring the structural changes during heating with high spatial resolution and precise awareness of location would provide valuable insights for understanding the overall thermal stability of the materials. High resolution transmission electron microscopy (HRTEM) is a suitable tool for probing the local details of the phase transformation during heating, because it offers both local structure and chemical information. [ 60 , 61 ]
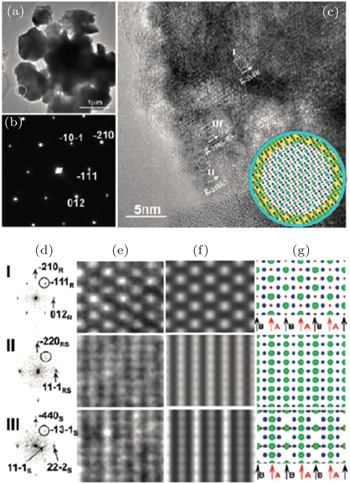
In our work, in situ TEM has been employed to study the structural origin of the overcharge-induced thermal instability of two cathode oxide materials that exhibit significant differences in thermal stability, NCA and NCM. [ 30 ] Detailed TEM analysis reveals that overcharged NCA and NCM particles both have complex core-shell-surface structures, which cannot be detected by XRD. For overcharged NCA, HRTEM imaging reveals three structures in the scale of tens of nanometers, as shown in Fig.
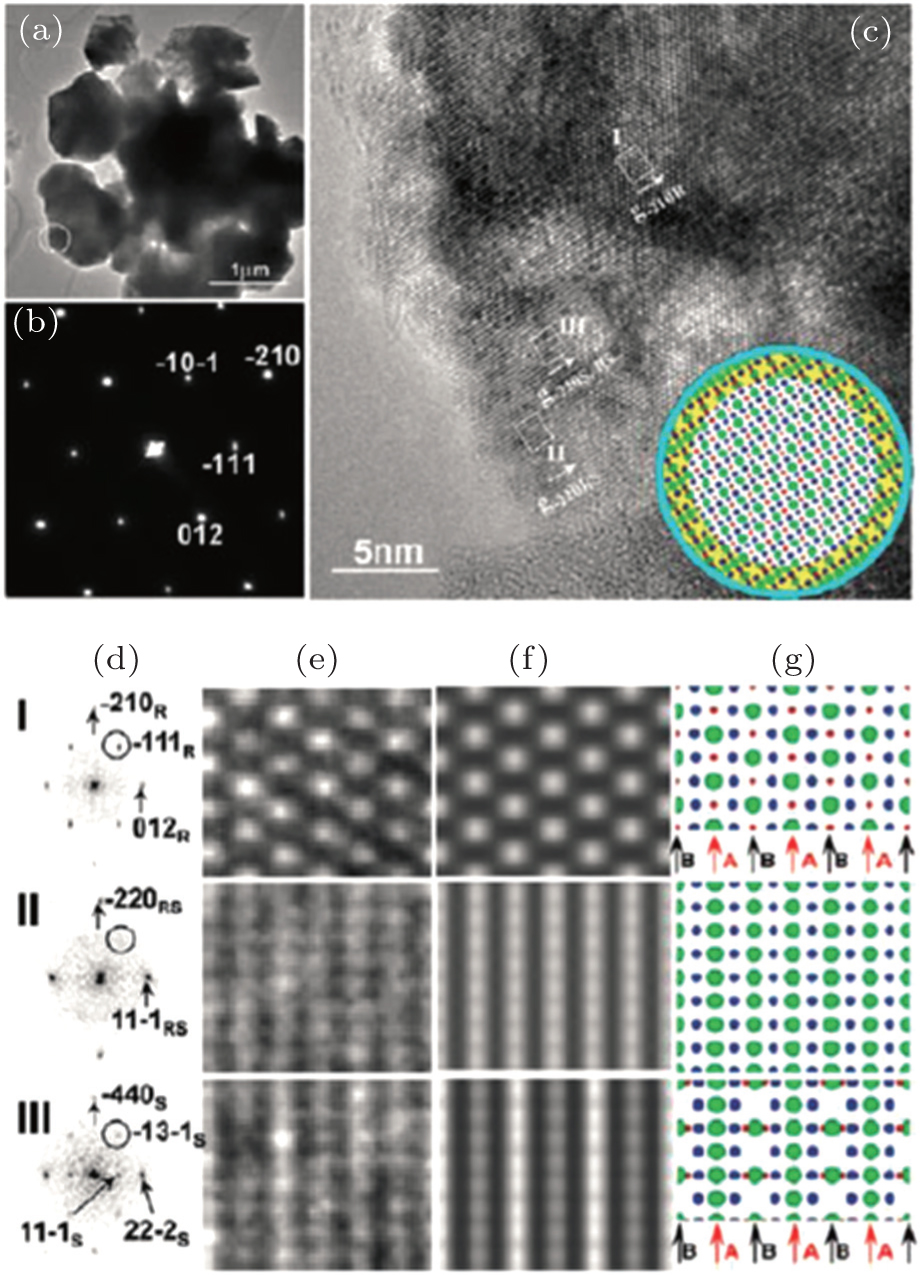 | Fig. 9. HRTEM images, selected area electron-diffraction pattern (SAEDP) and simulated structure of overcharged Li x Ni 0.8 Co 0.15 O 2 . Three phases, rhombohedral (in the core), spinel (in the shell), and rock-salt (on the surface), have been identified on the charged particle in the same image. [ 30 ] |
Note that structural decomposition has been found on the surface of the overcharged particle at room temperature, as mentioned above. For Li x Ni 0.8 Co 0.15 O 2 ( x < 0.15), the overcharge-induced new phase propagates from the surface to the bulk of particles, following the process of rhombohedral to spinel to rock-salt. This resembles the phase transformation sequence observed in heating. For most transition metal oxide cathode materials, the structural decomposition, accompanied by oxygen-release, often occurs in a deeply delithiated state (high voltage charging). This kind of structural transformation normally proceeds mildly and starts from the surface of the material, making it difficult to track by bulk characterization tools such as XRD. However, understanding the origin of the structural decomposition is vital to the development of stable cathode materials, because these subtle irreversible structural changes will accumulate during electrochemical cycling and result in capacity fading during long-term cycling. As revealed, the surface structure evolution during electrochemical cycling is similar to the bulk structure evolution during heating. The knowledge gained from thermal studies might provide valuable information for predicting the structural evolution of the material over long-term electrochemical cycling. This is understandable, because the metastable highly delithiated (or overcharged) structure tends to transform into a stable structure during electrochemical cycling, and heating accelerates this transformation. Therefore, approaches that are effective to improve the thermal stability of the material could also be applied to enhance its structural stability during electrochemical cycling. Recent research on lithium rich manganese based layered oxide cathode materials demonstrates this point. The lithium rich materials attract lots of interest nowadays, because they can deliver exceptionally high reversible capacity, exceeding 250 mA·h·g −1 between 4.8 V and 2 V. [ 54 , 63 , 64 ] Despite delivering high capacity, lithium-rich materials exhibit several practical shortcomings, such as continuous fading of both capacity and voltage during electrochemical cycling. [ 65 – 67 ] The scientific community has made substantial efforts to understand these phenomena. The layered-to-spinel structural transformation, accompanied by oxygen release, is found to occur on the surface of the material during charging at high voltage. This phase transformation behavior, which is considered to be one of the primary factors responsible for the voltage and capacity fading of lithium-rich layered material, is analogous to its thermal decomposition behavior at an early stage of heating (< 250 °C, results will be reported in our future work). [ 68 , 69 ] Surface coating, [ 70 ] an effective approach to improve thermal stability for most oxide materials, can also be applied to suppress the oxygen-release of lithium-rich materials during high voltage charging and to retard the structural transformation into the spinel phase. Strategies of substituting specific cations that are able to inhibit the thermal decomposition of the material during heating [ 67 ] can also be applied to alleviate the structural transformation of lithium-rich layered material during electrochemical cycling. Bear in mind that the structural evolution revealed by thermal studies is more significant and easier to identify than the subtle structural changes occurring during each electrochemical cycle. Thermal studies may provide an alternative way to understand the structural origin of the cyclic instability of the materials and offer useful guidance in developing more structurally stable electrode materials for lithium ion batteries.
The structure changes of several typical charged oxide cathode materials (NCA, NCM, and high voltage spinel) during a heating process, at both bulk and atomic levels, are reviewed in a comparative way, based on results obtained from in situ time-resolved XRD and MS, in situ XAS, and in situ TEM experiments. It has been found that the structural transformation (or decomposition) together with oxygen-release is inevitable for charged oxide cathode materials during the heating process. Several approaches are proposed to improve the thermal stability of oxide cathode materials: (i) substituting specific cations that require only slight reduction to migrate into tetrahedral sites of the oxygen framework at early stages of heating is an effective way to improve the intrinsic thermal stability of the material. On one hand, the slight reduction of these cations will generate only a small amount of oxygen. On the other hand, the occupation of these ions in the tetrahedral sites will impede the migration of other transition metal ions that are required for further structural transformation, so the onset temperature of structural transformation will be pushed higher. (ii) Surface modification that can prevent the structure decomposition from the surface is another effective approach to improve the thermal stability of the cathode materials. In addition, it has been revealed that the structural transformation of the oxide cathode materials, due to high voltage charging during electrochemical cycling, is similar to their structural transformation observed during heating. Therefore, the information obtained from thermal studies may also provide valuable insights for developing electrode materials with better cycle stability.
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 | |
| 23 | |
| 24 | |
| 25 | |
| 26 | |
| 27 | |
| 28 | |
| 29 | |
| 30 | |
| 31 | |
| 32 | |
| 33 | |
| 34 | |
| 35 | |
| 36 | |
| 37 | |
| 38 | |
| 39 | |
| 40 | |
| 41 | |
| 42 | |
| 43 | |
| 44 | |
| 45 | |
| 46 | |
| 47 | |
| 48 | |
| 49 | |
| 50 | |
| 51 | |
| 52 | |
| 53 | |
| 54 | |
| 55 | |
| 56 | |
| 57 | |
| 58 | |
| 59 | |
| 60 | |
| 61 | |
| 62 | |
| 63 | |
| 64 | |
| 65 | |
| 66 | |
| 67 | |
| 68 | |
| 69 | |
| 70 |