2.2.1.Numerical descriptorsThe re-annotation program was developed by integrating the TN curve and the Z curve methods[4] (for details, please refer to www.cbi.seu.edu.cn/RPGM). To numerically display the intrinsic properties that differentiate between protein-coding and non-coding sequences, 75 numerical parameters were derived from the two kinds of graphical representations.
The TN curve can project 64 kinds of trinucleotides into a two-dimensional space, [5] and then each trinucleotide is numerically decided by coordinates (x, y) on the basis of which z = x * y is defined. Based on the encoding strategy of the TN curve, 54 character parameters corresponding to the geometric center of each variable are derived to exhibit the specific property of composition and order of trinucleotides along the DNA sequences, which is written as Eqs. (1), (2), and (3).

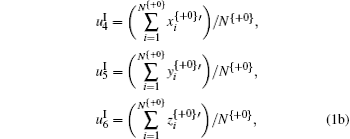


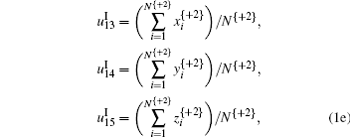







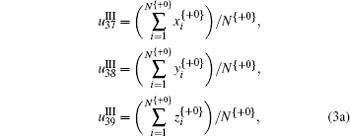
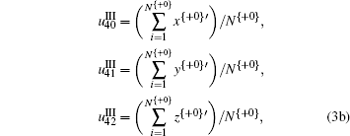



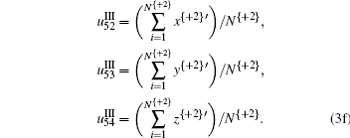
It is noted that in Eq. (3), + 0, + 1, and + 2 represent the three forward-reading frames from 5′ to 3′ of each sequence, respectively, and N{} is the total number of trinucleotides in each reading frame; I, II, and III denote three encoding strategies to decide (x, y), which is dependent on the first, second, and third base category, respectively; x’ , y′ , and z′ display the cumulative effect of x, y, and z, respectively.
In contrast to the TN curve, the 21 parameters derived from the Z curve exhibit the statistical properties of each codon position, [9] including nine position-specific parameters (Eq. (4)):

where i = 1, 2, 3 denotes the three codon positions. Besides this, 12 phase-specific dinucleotides are also illustrated by the following Z-transform (Eq. (5)):
 | (5) |
In Eq. (5), X = A, C, G, T; p(AA), p(AC), … , p(TT) represent the occurring frequencies of the 16 dinucleotides AA, AC, … , TT, respectively.
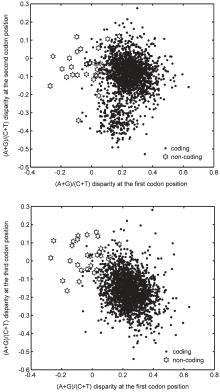
As is well known, the intrinsic difference between protein-coding genes and non-coding sequences lies in the former having regularly specific features, such as asymmetric nucleotide distributions at the three codon positions and codon usage bias, whereas the latter does not have such features. Thus, the ability to propose efficient numerical descriptors to exhibit the specific features of protein-coding genes is the core for gene prediction programs. In this work, 54 numerical descriptors derived from the TN curve and 21 statistic numerical descriptors derived from the Z curve method were used to demonstrate the specific features of protein-coding genes. As has been introduced before, [4, 10, 11] the numerical descriptors derived from the TN curve can exhibit information about the composition and distribution of trinucleotides along DNA sequences. In contrast, the numerical descriptors derived from the Z curve provide statistical information about the base compositions at the three codon positions as well as the dinucleotide compositions.[9] Therefore, the two groups of parameters can complement each other, displaying comprehensive sequence properties of protein-coding genes from different angles. On the other hand, the differences between protein-coding genes and non-coding sequences are universal in all species, which has been the foundation of gene prediction algorithms. In this work, the 75 derived parameters were used to demonstrate these universal properties that differentiate protein-coding sequences from non-coding sequences; therefore, it is conceivable that the present method is applicable to other prokaryotic genomes.







{kind=link}
{kind=link}
 , Sui Tian-Xiang
, Sui Tian-Xiang