2.1. ORR mechanisms Without considering any of the possible parasitic side reactions, the fundamental chemistry of an aprotic Li-O 2 battery during discharge undergoes these possible elementary reactions: [ 20 – 24 ]



To identify the ORR mechanisms in aprotic Li-O 2 cells, several research groups investigated the ORRs with multiple techniques including voltammetry, [ 20 – 23 ] electrochemical surface-enhanced Raman spectroscopy (EC-SERS), [ 24 – 26 ] differential electrochemical mass spectrometry (DEMS), [ 27 ] etc. For instance, Laoire et al . studied ORRs in various organic solvents (such as acetonitrile [ACN] and dimethylsulfoxide [DMSO]) containing various cations (such as tetrabutylammonium [TBA + ] and alkali metal ions of Li + , Na + , and K + ) using cyclic voltammetry. [ 20 , 21 ] Meanwhile, the reaction between  and Li + was also investigated by Peng et al. (Fig. 1 ). With the addition of Li + , the reduction peak appears at higher potentials, and with increasing Li + concentration, the magnitude of the new peak increases at the expense of the area under the original O 2 reduction peak, meaning that electrochemical reduction is followed by a chemical step. This subsequent chemical reaction severely depletes the amount of
and Li + was also investigated by Peng et al. (Fig. 1 ). With the addition of Li + , the reduction peak appears at higher potentials, and with increasing Li + concentration, the magnitude of the new peak increases at the expense of the area under the original O 2 reduction peak, meaning that electrochemical reduction is followed by a chemical step. This subsequent chemical reaction severely depletes the amount of  , leading to a shift of the potential toward higher voltages. These observations are consistent with the proposed ORR mechanisms illustrated by Eqs. ( 1 )–( 4 ), i.e., O 2 is first reduced to intermediate
, leading to a shift of the potential toward higher voltages. These observations are consistent with the proposed ORR mechanisms illustrated by Eqs. ( 1 )–( 4 ), i.e., O 2 is first reduced to intermediate  via a one-electron process (Eq. ( 1 )), the
via a one-electron process (Eq. ( 1 )), the  binds with Li + ions forming LiO 2 , an unstable intermediate (Eq. ( 2 )). Then, LiO 2 further transforms to Li 2 O 2 through either electrochemical reduction (Eq. ( 3 )) or chemical disproportionation (Eq. ( 4 )). Abraham et al . also used hard soft acid base (HSAB) theory to clarify this ORR process. According to the HSAB theory, alkali metal cations such as Li + ions, which represent hard acids, cannot efficiently stabilize
binds with Li + ions forming LiO 2 , an unstable intermediate (Eq. ( 2 )). Then, LiO 2 further transforms to Li 2 O 2 through either electrochemical reduction (Eq. ( 3 )) or chemical disproportionation (Eq. ( 4 )). Abraham et al . also used hard soft acid base (HSAB) theory to clarify this ORR process. According to the HSAB theory, alkali metal cations such as Li + ions, which represent hard acids, cannot efficiently stabilize  , a relatively soft base. Hence, the unstable intermediate LiO 2 is prone to disproportionation, forming Li 2 O 2 , because
, a relatively soft base. Hence, the unstable intermediate LiO 2 is prone to disproportionation, forming Li 2 O 2 , because 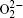 is a strong base. [ 20 , 21 , 28 ]
is a strong base. [ 20 , 21 , 28 ]
The proposed ORR mechanisms (Eqs. ( 1 )–( 4 )) were investigated by Peng et al . using EC-SERS. [ 24 ] With this spectroscopy-based technique, the reaction products and intermediates, which are crucial to the formulation of the ORR mechanisms in the aprotic Li + electrolyte, were identified directly. As shown in Fig. 2 , at the initial stage of ORRs, two newly formed species, LiO 2 (peak 2) and Li 2 O 2 (peak 3), can be identified; they are absent at open circuit potential, where no ORR takes place. Moreover, with the elapse of time, the signal associated with LiO 2 gradually decreases and vanishes, and only the signal of Li 2 O 2 remains. These observations clearly demonstrate that the unstable LiO 2 can transform to Li 2 O 2 via disproportionation, supporting the reaction mechanisms of either Eqs. ( 1 ), ( 2 ), and ( 4 ) or Eqs. ( 2 ) and ( 4 ).
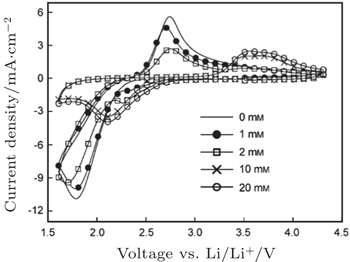
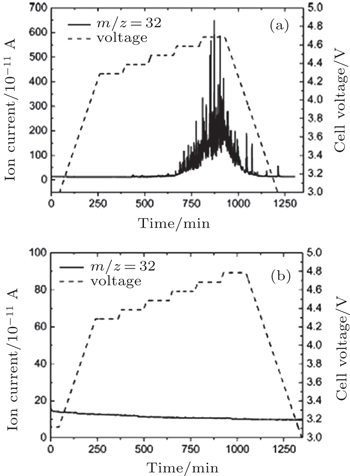
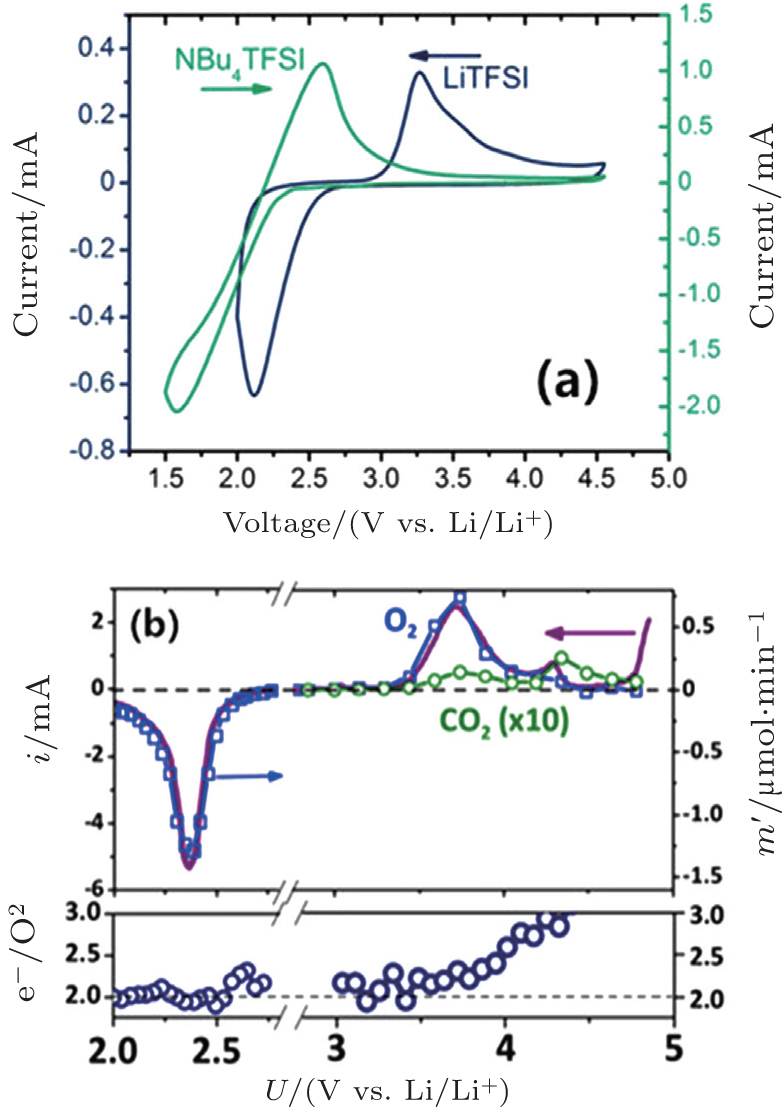
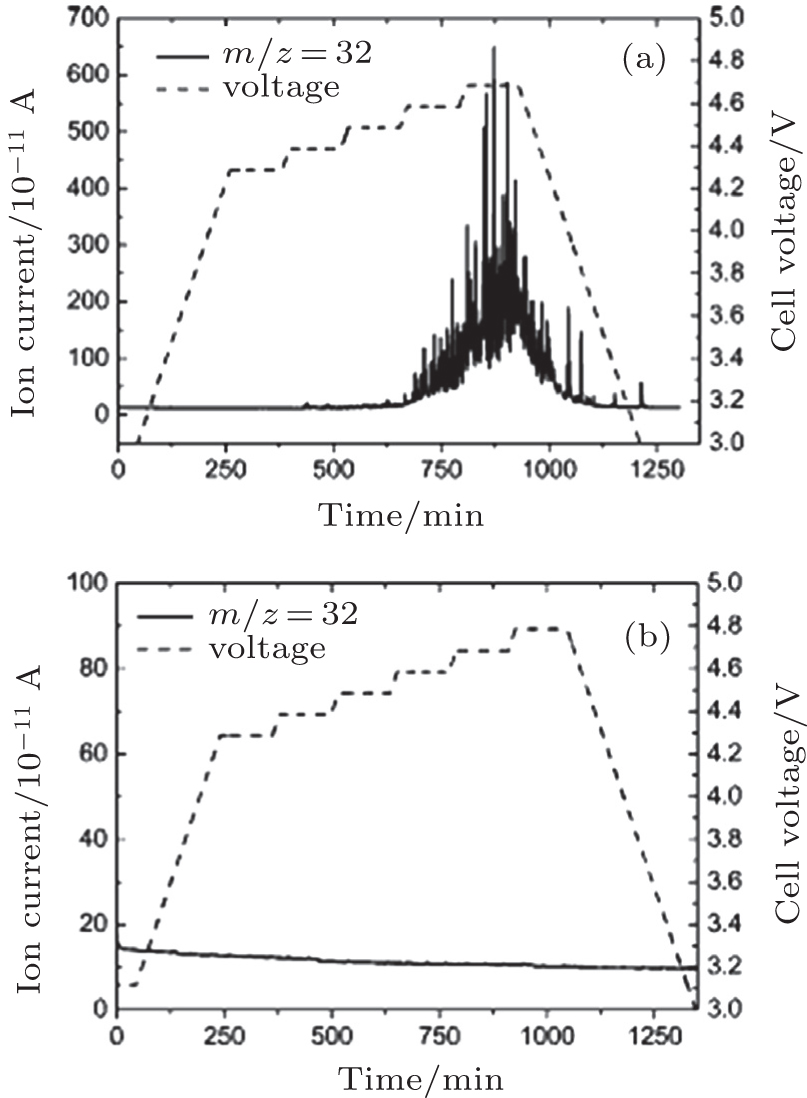
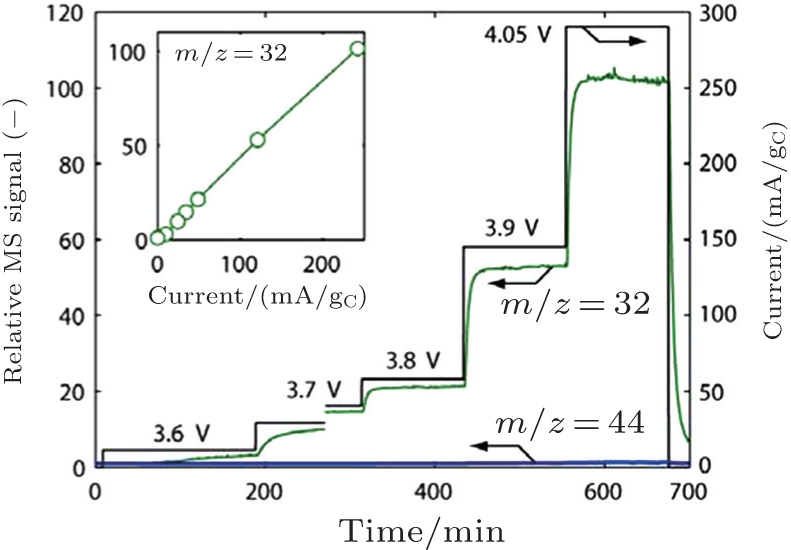
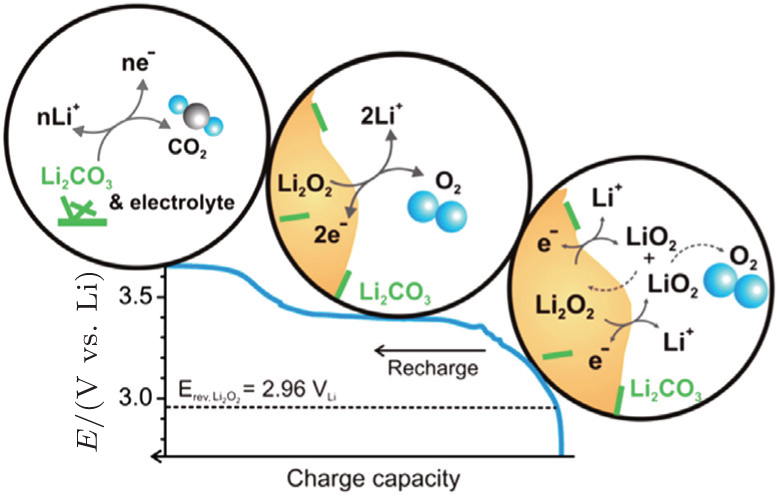
In addition, McCloskey et al . employed cyclic voltammetry (CV) and differential electrochemical mass spectrometry (DEMS) to study the oxygen reactions in aprotic Li-O 2 cells with a lithium bis(trifluoromethanesulfonyl)imide(LiTFSI)-dimethoxyethane (DME) electrolyte. [ 27 ] Interestingly, only one peak for ORRs was observed (Fig. 3(a) ). Their observations differ from many published results that demonstrated multiple peaks of ORRs in the cyclic voltammograms (CVs) and those peaks were attributed to one-electron processes. [ 20 , 21 ] McCloskey et al . argued that the extra reduction peaks in CVs are due to the presence of impurities, e.g., H 2 O. Moreover, the ∼2e – /O 2 process forming Li 2 O 2 has been confirmed by DEMS experiments, as exhibited in Fig. 3(b) . This means that once the LiO 2 is formed, it quickly transforms to Li 2 O 2 by reaction (Eq. ( 3 )) or reaction (Eq. ( 4 )), and it cannot be thermodynamically stable, contrary to the suggestions of others. [ 20 , 22 – 24 ]
Early CV studies of ORRs in aprotic solvents reported by Laoire et al . are excellent, but the reaction products and intermediates cannot be identified with certainty by these conventional electrochemical methods; [ 28 ] therefore, the mechanisms that were proposed based on CV measurements are questionable. The extra peaks of ORRs in CVs may be due to the existence of H 2 O in the electrolyte, as suggested by a recent work, [ 29 ] and the effects of residual H 2 O (or any proton sources) need further clarification. By EC-SERS, the ORR intermediate LiO 2 has been detected directly, which is very beneficial for the formulation of the ORR mechanisms. Spectroscopy-based techniques will continue to play a key role in better understanding the ORRs in various aprotic Li-O 2 batteries that incorporate a broad range of aprotic electrolyte solvents. However, DEMS experiments demonstrate that an overall two-electron process is obtained within the time resolution (seconds) of this technique, which means that LiO 2 may not be a stable intermediate and could rapidly transform to Li 2 O 2 . This contradiction between the DEMS and SERS results needs further study. So far, there is no consensus on the path of LiO 2 transformation to Li 2 O 2 under the conditions of Li-O 2 cell operation, i.e., whether it is dominated by electrochemical reduction or chemical disproportionation. Further investigations are urged, seeking a better understanding of ORRs in aprotic Li-O 2 batteries.
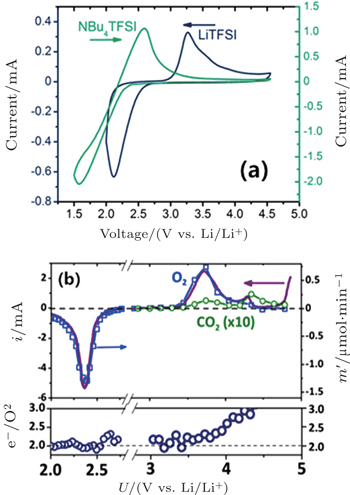
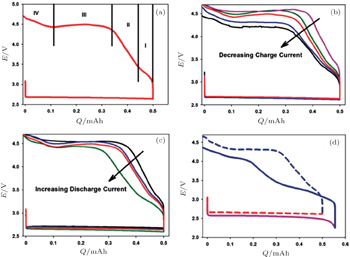
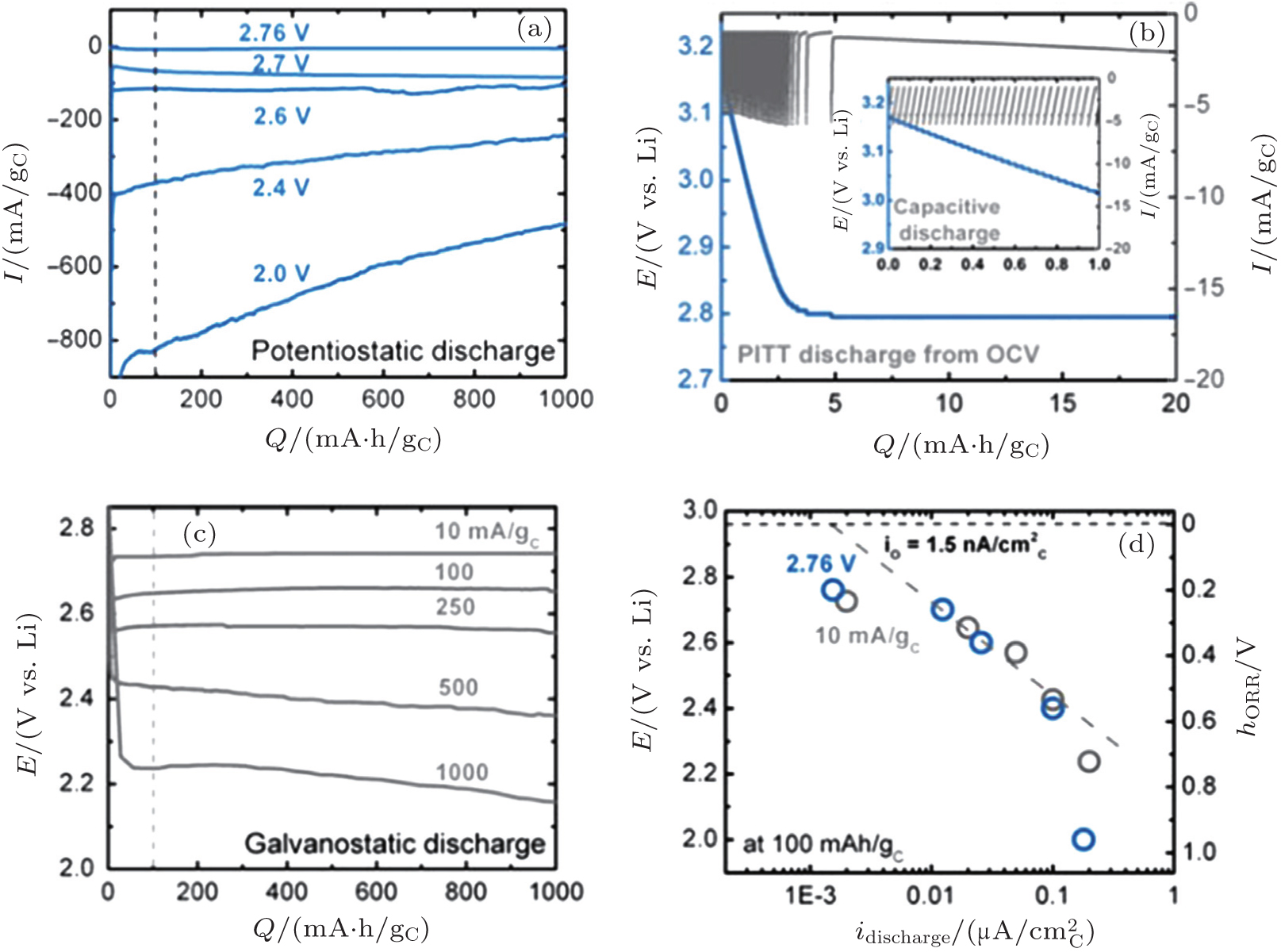
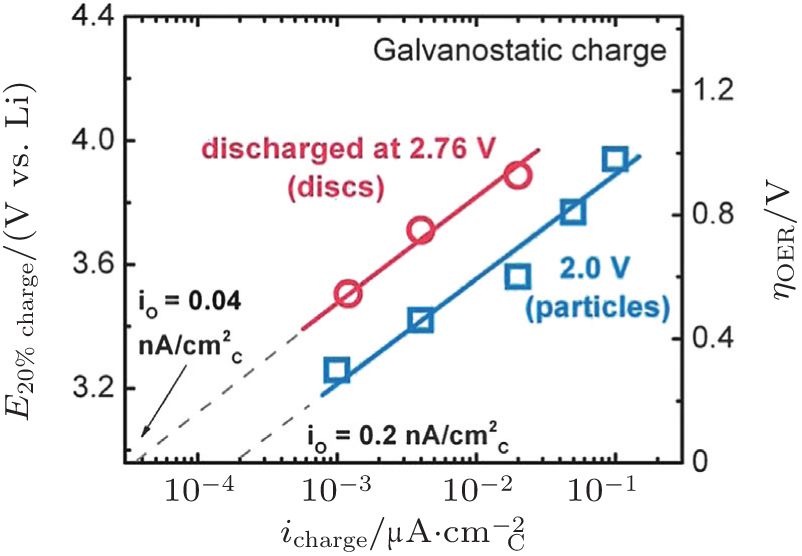
2.2. ORR kinetics Understanding the intrinsic ORR kinetics and transport limitations associated with Li 2 O 2 formation is crucial to the development of aprotic Li-O 2 batteries with the desired rate capabilities. To explore ORR kinetics, Gallant et al . investigated the ORR over-potentials on a carbon nanotube-based cathode by using galvanostatic and potentiostatic intermittent titration (PITT) techniques. [ 30 ] Under potentiostatic conditions, the average current increases in magnitude as the voltage is reduced from 2.76 V to 2.0 V (Fig. 4(a) ). The acquired 2.76 V is the maximum potential at which nucleation of Li 2 O 2 on CNTs occurs, corresponding to the PITT discharge at open circuit potential, as shown in Fig. 4(b) . These results were further validated by a galvanostatic discharge test (Fig. 4(c) ). Moreover, the discharge potential plateaus present a linear relationship with log i galvano , described in a Tafel curve (Fig. 4(d) ), confirmed with a kinetically controlled ORR procedure. Notably, the measured Tafel slope differs from the reported results for other types of cathodes. [ 31 ] Although the physical origin of this difference is not understood, the different Tafel slopes observed on various carbon surfaces at low Li 2 O 2 coverage may reflect different nucleation kinetics of Li 2 O 2 .
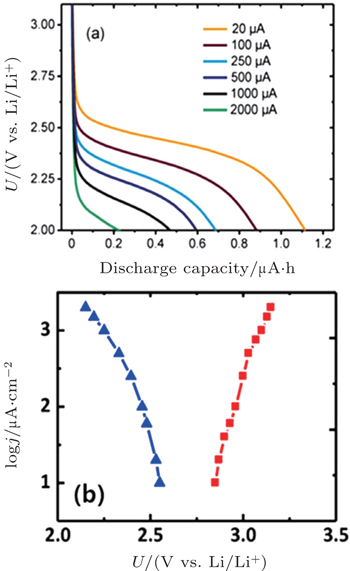
Many studies of discharge behavior and kinetic process using typical Swagelok-type cells with carbon cathodes were reported in the literatures, [ 30 , 31 ] however, Viswanathan et al . argued that these results may fully mask the fundamental kinetic behavior due to the cell’s intrinsic impedance. It is critical to eliminate the cell iR drop for measuring the fundamental kinetic over-potential. Therefore, Viswanathan and associates estimated the kinetic over-potentials by using a bulk electrolysis cell with a flat, polished, small-surface glassy carbon (GC) electrode. [ 32 ] Figure 5(a) shows galvanostatic discharge plots of Li-O 2 cell at various current densities i in the bulk electrolysis cell. The initial drop in potential is attributed to the kinetic over-potential, a linear decrease in potential with Q dis is ascribed to an iR drop through the thickening Li 2 O 2 film on the GC surface, and a “sudden death” is characterized by a rapid decrease in U . These authors discussed only the initial drop of potential U and its i dependence to probe the real kinetic behavior, because charge transport limitations for Li 2 O 2 growth dominate the electrochemistry during the later discharge. Figure 5(b) is the Tafel plot for the cell’s cycle, wherein it is plain to see that the kinetic over-potentials for discharge and recharge procedures are extremely small. These results imply that the electrical efficiency of a Li-O 2 battery is high in a discharge-recharge cycle if the battery is limited only by the kinetic over-potential.
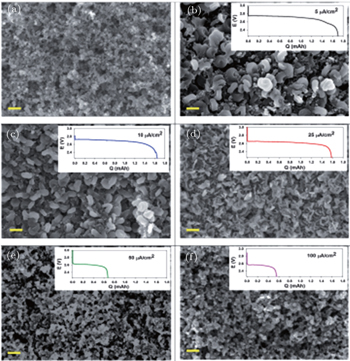
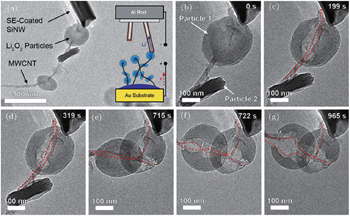
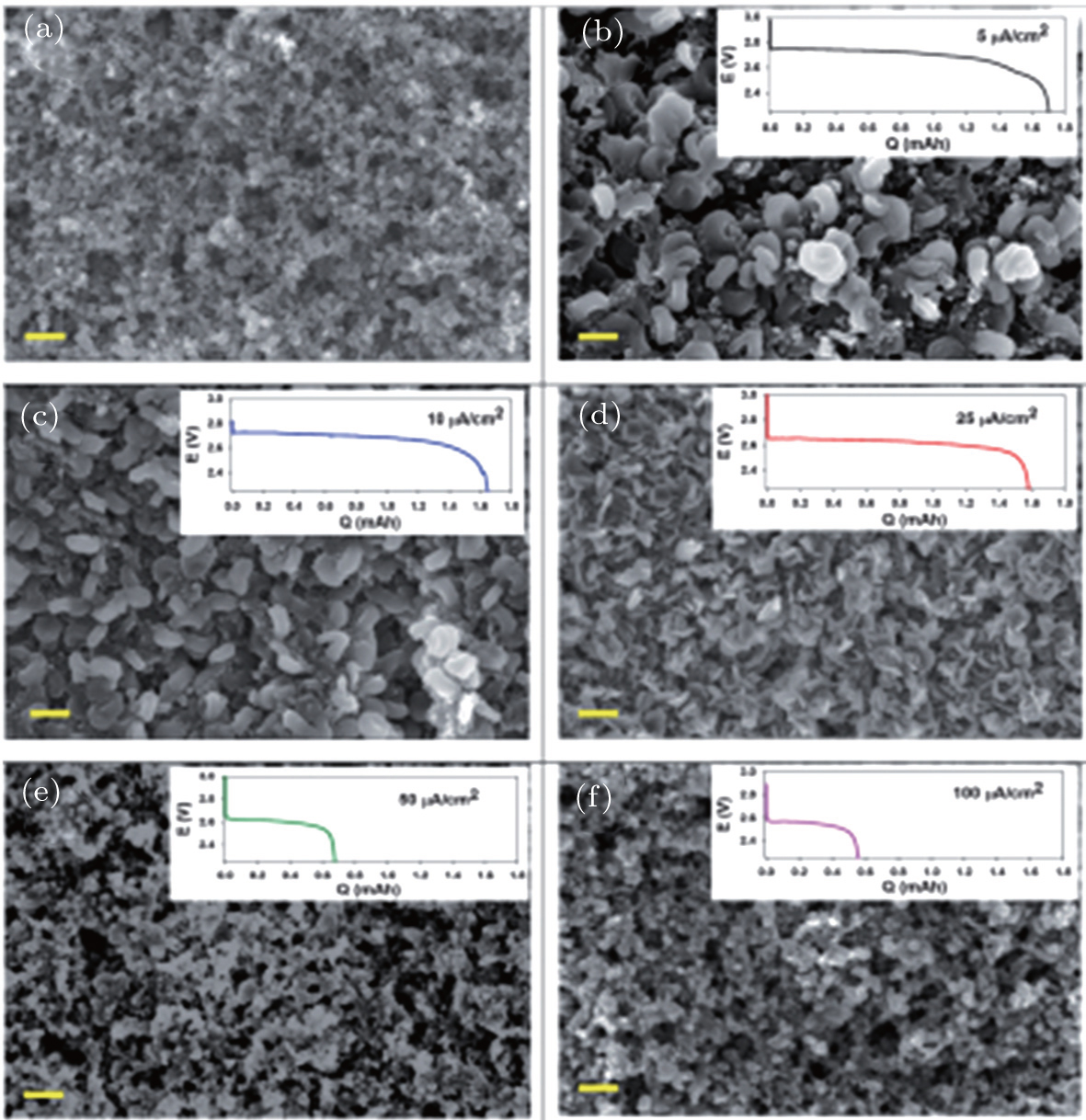
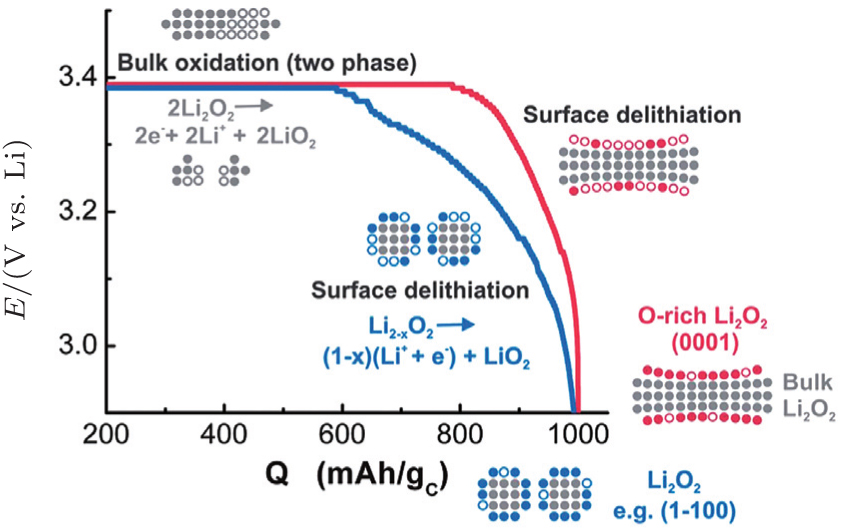
2.3. Li 2 O 2 morphology The reported morphology of Li 2 O 2 formed on a cathode is also controversial. Viswannathan et al . reported homogeneous films of Li 2 O 2 deposited on a small-surface GC cathode in an electrolysis cell, observed by atomic force microscopy (AFM). [ 33 ] Adams et al . also observed the formation of a quasi-amorphous peroxide film at high current rates, as shown in Figs. 6(e) and (f). [ 16 ] However, at low current rates, toroid-shaped Li 2 O 2 products have been found (Figs. 6(b) – 6(d) ) on a pristine carbon cathode (Fig. 6(a) ). Obtaining such small, poorly crystalline particles at high current density was ascribed to the direct second electro-reduction pathway (Eq. ( 3 )) due to the stronger adsorption of LiO 2 onto the Li 2 O 2 substrate at this high current rate. [ 16 ]
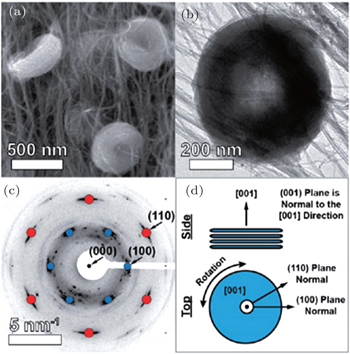
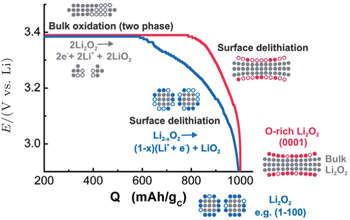
Other researchers also reported that the toroid-like Li 2 O 2 particles are formed on various types of carbon cathodes, especially at low discharge current density. [ 30 , 34 , 35 ] For instance, Shao-Horn and colleagues revealed the morphology evolution of these toroid-like products by using transmission electron microscopy (TEM), the formation originally began with the nucleation of small particles on the side wall of CNT and evolved upon continued discharge. The TEM investigation also showed that these toroids are highly crystalline with the Li 2 O 2 (0001) facet normal to the axis of the toroid as shown in Fig. 7 . It is worth noting that this observation of toroids has triggered much controversy about ORR mechanisms in aprotic Li-O 2 batteries, because the morphology is totally different from the films observed in an electrolysis cell, and the toroid-like Li 2 O 2 particles with poor electrical conductivity and large size (several hundred nanometers) can be electrochemically oxidized. In a recent work, the formation of the Li 2 O 2 toroids was ascribed to the existence of H 2 O impurity, [ 29 ] i.e., H 2 O induces some solubility of LiO 2 due to its high acceptor number, which helps validate the solution mechanism for Li 2 O 2 formation proposed by others. [ 16 ]















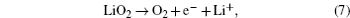
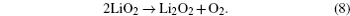


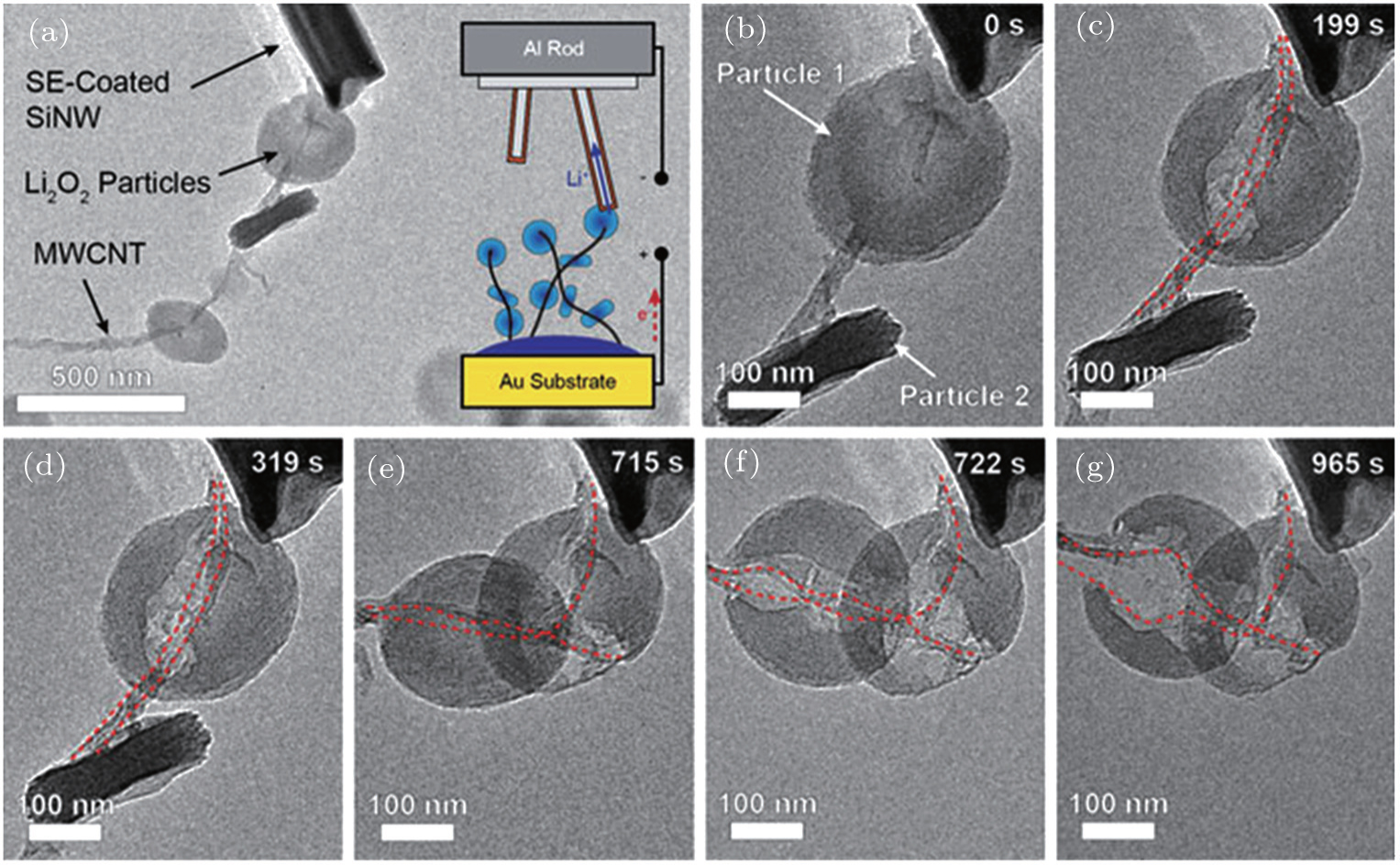
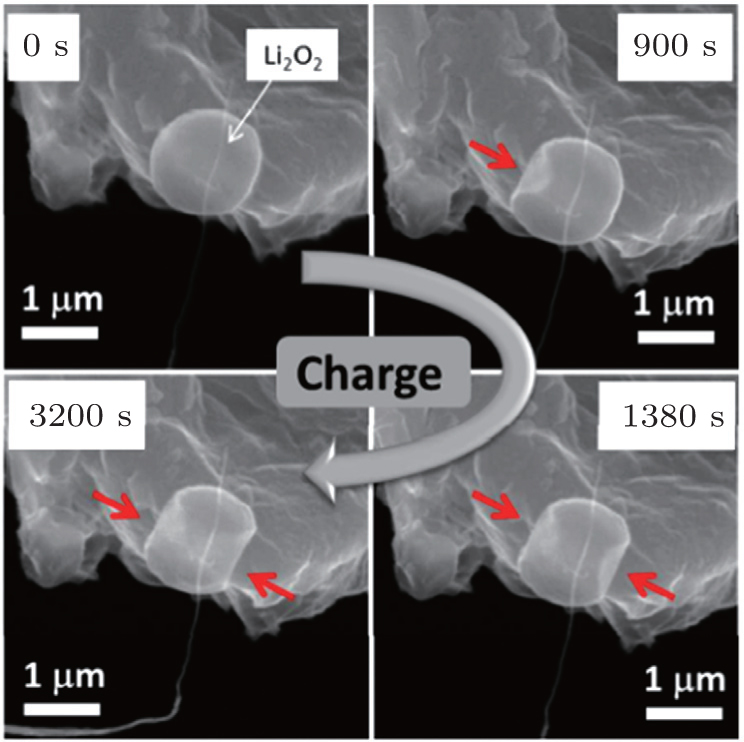

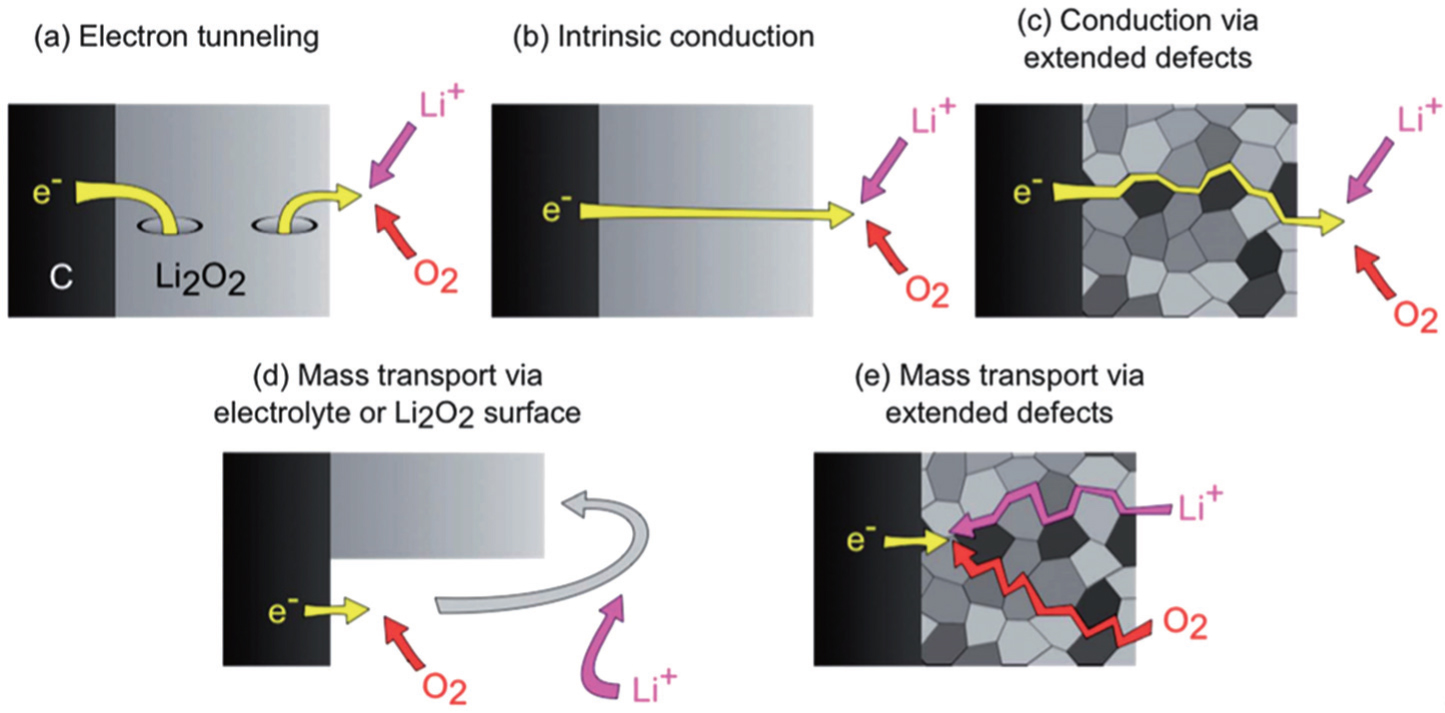

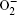
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 , Zhang Yelong 1, 2, †,
, Zhang Yelong 1, 2, †,