



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Dependence of mechanical properties on the site occupancy of ternary alloying elements in γ′-Ni3Al: Ab initio description for shear and tensile deformation
Cite this Article
Wen Minru, Xie Xing, Dong Huafeng, Wu Fugen, Wang Chong-Yu. Dependence of mechanical properties on the site occupancy of ternary alloying elements in γ′-Ni3Al: Ab initio description for shear and tensile deformation. Chinese Physics B, 2020, 29(7): 078103 
Permissions
Dependence of mechanical properties on the site occupancy of ternary alloying elements in γ′-Ni3Al: Ab initio description for shear and tensile deformation
† Corresponding author. E-mail:
Project supported by the National Natural Science Foundation of China (Grant No. 11804057), the Natural Science Foundation of Guangdong Province, China (Grant No. 2017B030306003), and the National Key Research and Development Program of China (Grant No. 2017YFB0701500).
Abstract
The site occupancy behavior of ternary alloying elements in γ′-Ni3Al (a key strengthening phase of commercial Ni-based single-crystal superalloys) can change with temperature and alloy composition owing to the effect of entropy. Using a total-energy method based on density functional theory, the dependence of tensile and shear behaviors on the site preference of alloying elements in γ′-Ni3Al were investigated in detail. Our results demonstrate that Fe, Ru, and Ir can significantly improve the ideal tensile and shear strength of the γ′ phase when occupying the Al site, with Ru resulting in the strongest enhancement. In contrast, elements with fully filled d orbitals (i.e., Cu, Zn, Ag, and Cd) are expected to reduce the ideal tensile and shear strength. The calculated stress–strain relationships of Ni3Al alloys indicate that none of the alloying elements can simultaneously increase the ideal strength of the γ′ phase for both Ni1-site and Ni2-site substitutions. In addition, the charge redistribution and the bond length of the alloying elements and host atoms during the tensile and shear processes are analyzed to unveil the underlying electronic mechanisms.
Keyword:;stress-strain relations;;transition-metal elements;;γ′-Ni3Al;;first-principles calculations;
1. Introduction
As a key strengthening phase of commercial Ni-based single-crystal superalloys, the ordered γ′-Ni3Al precipitate phase is of great technological importance owing to its excellent elevated-temperature creep and oxidation resistance as well as the anomalous temperature dependence of the yield stress.[1–5] The stoichiometric γ′-Ni3Al L12 structure with space group 
Various experimental methods can be used to explore the site occupancy of alloying elements in Ni3Al, including x-ray diffraction, transmission electron microscopy, and atom probe methods.[15–21] However, these experimental studies are costly, and only some transition-metal (TM) elements are considered. Theoretical research on the site preference of alloying elements can make up for the deficiencies of current experimental research methods. Early theoretical investigation was mainly based on the phenomenological Ising-type models.[22–24] For example, Wu et al.[23] studied the site preference in ordered L12-Ni3Al and observed that the temperature and alloy composition affect the occupancy behavior of alloying elements. Ruban and Skriver[12,25] studied the site occupation of 3d, 4d, and 5d TM elements in the γ′ phase using the coherent potential approximation in conjunction with the linear muffin-tin orbitals method. The currently popular theoretical method to calculate the site substitution behavior in ordered compounds is first-principles density functional theory (DFT) calculations using the Wagner–Schottky model.[13,14,18,26–31] By adopting this method, our previous calculations[29] of the site occupancy of TM elements in Ni3Al indicated that the site behaviors of Fe, Co, Cu, Zn, Ru, Rh, Ag, Cd, and Ir are strongly related to the alloy composition at T = 0 K.
Similar to elastic constants, the ideal strength (the minimum stress required to yield a defect-free crystal) of a solid material is a primary intrinsic parameter for understanding the mechanical behaviors of solid structural materials. However, unlike the extensive theoretical efforts devoted to investigating the effect of substitutional TM elements on the elastic properties of the γ′-Ni3Al phase, the study of the TM-element dependence of the ideal strength of the γ′ phase is limited. For L12-Ni3Al, the weakest uniaxial tensile direction is the 〈 110〉 direction, and the weakest shear slip system is the 

In light of this background, the key purpose of the present research is to investigate the effect of Fe, Co, Cu, Zn, Ru, Rh, Ag, Cd, and Ir additions on the ideal shear and uniaxial tensile strengths with different site occupancies in γ′-Ni3Al using ab initio calculations. In addition, the charge redistribution and the chemical bonding between the alloying elements and host atoms in the tensile and shear processes were analyzed to unveil the underlying electronic mechanism.
2. Computational methods
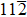
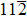

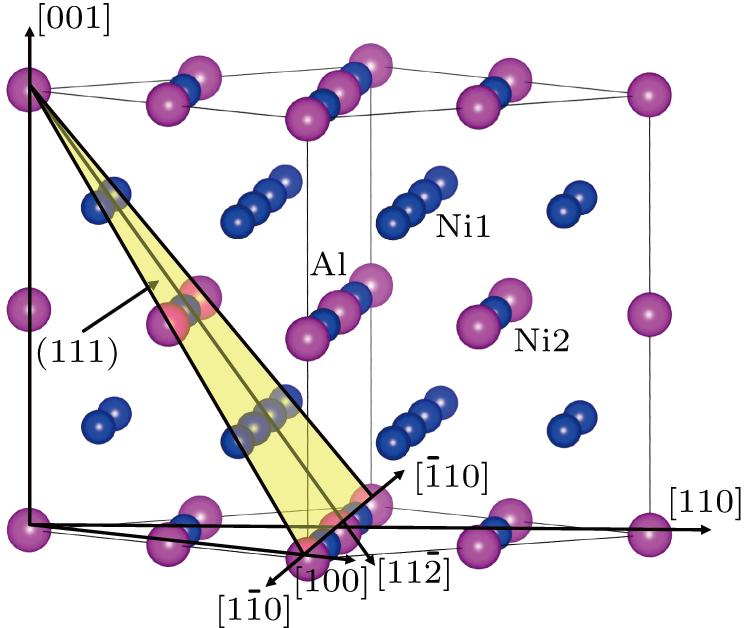
In this work, we studied tension in the [110] direction (the weakest uniaxial tensile orientation) and shear in the 
 | Fig. 1. Schematic illustration of Ni3Al 32-atom 2× 2× 2 supercell. The Al and Ni atoms are denoted by purple and blue balls, respectively. The yellow shadow area represents the (111) slip plane. |
By adopting Wagner–Schottky model,[41] our previous study[29] calculated the transfer energy 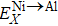
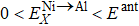
To simulate the shear and tensile processes, we adopted the standard approach described in Refs. [26,27]. For [110] uniaxial tension, we set the loading direction along the x axis. Hence, the deformed lattice vector R is given by R = R0 D, where R0 is the primitive non-deformed vector, and the tensile deformation matrix Dtension is

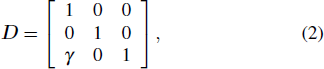


In the present study, the lattice vector was deformed from 0.0 to 0.18 and 0.28 in steps of 0.02 for the tensile and shear process, respectively. The structures of all the calculated systems were completely relaxed until reaching the force and energy criteria at each deformed strain. The stress–strain relationship was obtained from the calculated tensile and shear energies using Eqs. (
The DFT calculations were performed using the Vienna Ab Initio simulation package (VASP)[49] with projector augmented-wave (PAW) pseudopotentials[50] for all calculations. The exchange–correlation functional was described by the Perdew–Burke–Ernzerhof (PBE) generalized gradient approximation (GGA).[51] A Monkhorst–Pack[52] k-mesh of 11× 11× 11 was adopted in this work, and the plane-wave kinetic energy cutoff was set to 400 eV. The crystal structure was relaxed until the force converged to less than 0.01 eV/Å, and the energy convergence for electronic self-consistency was 10−5 eV.
3. Results and discussion
Dominated by the behavior of chemical bonding, common slip, and nucleation of dislocations,[53–55] the ideal strength of solid materials sets the upper bound of the strength achievable by a real material under certain loading conditions. Investigations of the ideal strength are fundamental to understand the behavior of fracture, failure,[56–58] and creep[59,60] for advanced materials as well as to determine the gap between the real strength of materials and their ideal strength.[61] Ab initio computational [110] tensile and 
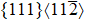
| Table 1. Calculated ideal strengths (in unit GPa) and corresponding strains of Ni3Al alloys doped with alloying element, along with other DFT predicted values. . |
The tensile and shear behavior of Xs-doped Ni3Al, where Xs denotes the TM elements X (X = Fe, Co, Cu, Zn, Ru, Rh, Ag, Cd, and Ir) occupying the s site (s = Al, Ni1, and Ni2 sites), were systematically studied in this work. The calculated ideal strengths for all the substitutional systems are summarized in Table
When TM elements X occupied the Al site, according to Table 
Compared with Al-site defected alloys, Ni-site defected Ni3Al results in a different scenario. There are two non-equivalent Ni sites in γ′-Ni3Al: the Ni1 site of the [


To better illustrate the effect of the ternary alloying additions on the ideal strength of Ni3Al alloys, the relations between ΔσFM and ΔτFM (both expressed in percent of the ideal strength of stoichiometric Ni3Al) are plotted in Fig. 
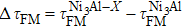
 | Fig. 2. Correlations between  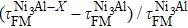 |
To obtain deeper insight into the underlying electronic mechanism responsible for the various efficiencies of ternary alloy element additions on the tensile and shear behavior, we investigated the charge redistribution induced by the substitutional impurities in γ′-Ni3Al during the tensile and shear processes. Figures 
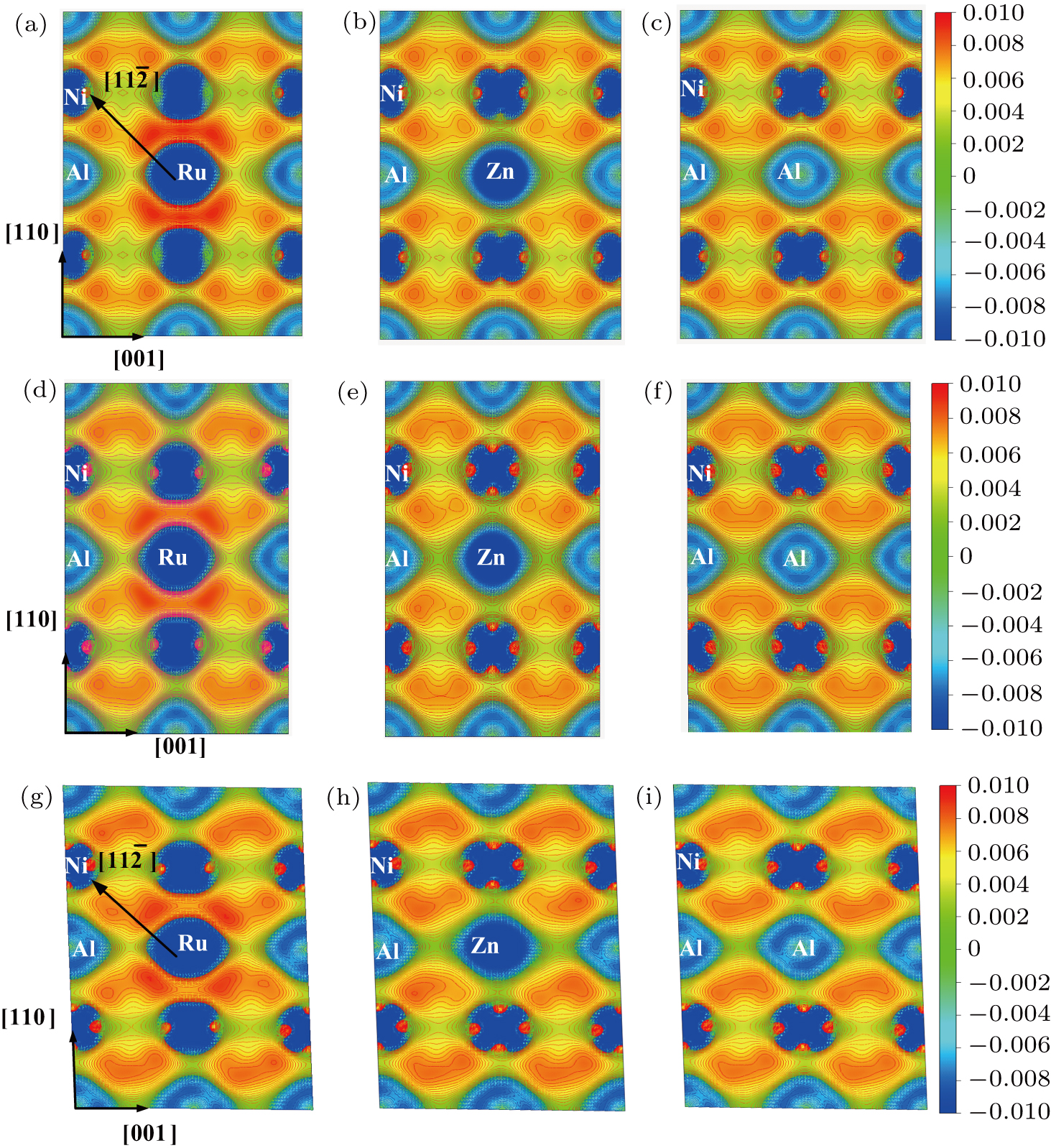 | Fig. 3. Charge density difference of ( |
As observed in Figs.
Figures 
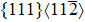
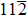
In terms of Ni-site defect alloys, the ideal tensile and shear strength are dependent on the ternary elements as well as the doping site. To reveal the strengthening mechanism, we also studied the variation of the bond lengths between the TM element X (X = Ru, Zn, and Ni) and its FNN host atoms in Ni23Al8Ru, Ni23Al8Zn, and stoichiometric Ni3Al during the uniaxial tensile processes. At zero strain, the impurity atom X possesses 12 FNN atoms consisting of 4 Al atoms, 4 Ni1 atoms, and 4 Ni2 atoms (see insets of Fig.
 | Fig. 4. Variation of bond lengths between the TM element X (X = Ru, Zn, and Ni) and its FNN host atoms normalized with respect to their respective equilibrium values during [110] tensile tests for Ni23Al8Ru, Ni23Al8Zn, and stoichiometric Ni3Al. The left (right) inset shows the substitutional impurity atom X and its FNN atoms for XNi1-doped (XNi2-doped) alloys at zero strain. The purple, blue, and green balls represent Al, Ni, and X atoms, respectively. The red dashed lines in the insets mark the shortest X–Ni bond during the tensile loading. |
As demonstrated in Fig.
4. Conclusions
The effects of Fe, Co, Cu, Zn, Ru, Rh, Ag, Cd, and Ir on the ideal shear and uniaxial tensile behaviors with different site occupancy in γ′-Ni3Al were studied in detail using ab initio total-energy methods. Our results indicate that the dependence of the ideal strength on the site preference of alloying element X in γ′-Ni3Al is associated with the d-shell occupancy of X. Elements with partially filled d orbitals (i.e., Fe, Co, Ru, Rh, and Ir) enhance the ideal tensile strength when occupying the Al site in Ni3Al, whereas elements with fully filled d orbitals (i.e., Cu, Zn, Ag, and Cd) lead to a decrease of τFM and σFM. Furthermore, the strengthening effect of Ru arises from the covalent-like bonds between impurities and host atoms according to analysis of the charge redistribution of Ni3Al alloys, whereas the detrimental effect of Zn is caused by the metallic bonds between Zn and Ni. In terms of Ni-site defect Ni3Al, elements with partially filled d shells are expected to weaken (improve) the ideal tensile and shear strength of the alloys when they occupy the Ni1 (Ni2) site, and the elements with fully filled d shells have the reverse effect. Moreover, the variations of the bond lengths between the TM element X (X = Ru, Zn, and Ni) and its FNN host atoms during uniaxial tensile tests indicate that Ru can effectively enhance (weaken) the deformation resistance and mechanical behaviors of the γ′ phase when occupying the Ni2 (Ni1) site, whereas the opposite is true for Zn. In addition, a linear relationship for ΔσFM versus ΔτFM was observed for the alloys with Al-, Ni1-, and Ni2-site defects in this study. Our findings provide insight into the reinforcement effects of ternary alloying elements on the ideal strength with different site preferences in γ′-Ni3Al, which have not been previously studied using first-principles calculations.
Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] | |
| [33] | |
| [34] | |
| [35] | |
| [36] | |
| [37] | |
| [38] | |
| [39] | |
| [40] | |
| [41] | |
| [42] | |
| [43] | |
| [44] | |
| [45] | |
| [46] | |
| [47] | |
| [48] | |
| [49] | |
| [50] | |
| [51] | |
| [52] | |
| [53] | |
| [54] | |
| [55] | |
| [56] | |
| [57] | |
| [58] | |
| [59] | |
| [60] | |
| [61] |