






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Structural, mechanical, and electronic properties of Zr – Te compounds from first-principles calculations
Cite this Article
Wang Peng, Zhang Ning-Chao, Jiang Cheng-Lu, Liu Fu-Sheng, Liu Zheng-Tang, Liu Qi-Jun. Structural, mechanical, and electronic properties of Zr – Te compounds from first-principles calculations. Chinese Physics B, 2020, 29(7): 076201 
Permissions
Structural, mechanical, and electronic properties of Zr – Te compounds from first-principles calculations
† Corresponding author. E-mail:
Project supported by the National Natural Science Foundation of China (Grant No. 11574254), the Key Research Project of Science and Technology Department of Shaanxi Province, China (Grant Nos. 2018GY-044 and 2017ZDXM-GY-114), the Innovation Talent Promotion Project of Shaanxi Province, China (Grant No. 2019KJXX-034), the Science and Technology Program of Sichuan Province, China (Grant No. 2018JY0161), and the Fund of the State Key Laboratory of Solidification Processing, Northwestern Polytechnical University, China (Grant No. SKLSP201843).
Abstract
The first-principles calculations based on density functional theory are used to obtain structural, mechanical, and electronic properties of Zr–Te compounds. The optimized structural parameters are consistent with the available experimental data. The calculated mechanical properties and formation energy show that the Zr–Te compounds are all mechanically and thermodynamically stable. The bulk modulus B, shear modulus G, Young’s modulus E, Debye temperature ΘD, and sound velocity vm are listed, which are positively correlated with the increasing of atomic fraction of Zr. The behaviors of density of states of Zr–Te compounds are obtained. Furthermore, the electronic properties are discussed to clarify the bonding characteristics of compounds. The electronic characteristics demonstrate that the Zr–Te systems with different phases are both covalent and metallic.
1. Introduction
Zr-based bulk amorphous alloys play a significant role in the history of alloys,[1] and have aroused the wide interest in glass-forming ability to become bulk metallic glasses.[2–5] The first synthesized metallic glasses were reported in 1960,[6] which contained zirconium. Zirconium is non-toxic and Zr-based alloys show good biocompatible ability, so that they can be used as biomedical materials.[7] Besides, Zr-based alloys possess wide applications in battery cathodes,[8] solar cells,[9] nuclear industry,[10] and transistors.[11] Recently, many researchers have investigated Zr–Te compounds in order to develop their advanced applications in superconductors, thermoelectric materials, spintronics/quantum computing, flexible electronics,[11–16] thus showing their huge potential applications in the electronic industry. Superconductivity of ZrTe3 was found under pressure by measuring the resistivity of single crystal, and no structural phase transition was observed.[12] The ZrTe5 was predicted to be a good thermoelectric material due to its large Seebeck coefficient and good electric conductivity.[13] Temperature-dependent resistivity and magneto-transport characterizations of polycrystalline ZrTe5 – δ have been reported,[14] indicating that it is a p-type semiconductor. In addition, the structural, elastic, electronic, and transport properties of semimetal ZrTe have been investigated.[15,16] It can be seen that different compositions of Zr and Te result in different physical properties and various applications. However, it is quite lacking in thorough and systematic investigations of Zr–Te compounds.
In order to offer a comparative study between Zr–Te compounds, the fundamental properties of ZrTe, ZrTe3, ZrTe5, Zr2Te, Zr3Te, Zr5Te4, and Zr5Te6[17–25] are investigated by using the first-principles calculations, which include structural, elastic, mechanical, and electronic properties.
2. Computational methods
All calculations were performed with CASTEP code[26] based on the density functional theory (DFT). The local density approximation (LDA) with the Ceperley–Alder–Perdew–Zunger (CA-PZ) functional[27] was employed as the exchange-correlation functional due to the fact that the LDA can better describe the electronic characteristics of metals. Using plane-wave expansions, the wave functions of Kohn–Sham[28] were expanded. The Monkhorst–Pack k-points were used to optimize the structures for the sampling of Brillouin zone, including 3 × 7 × 4, 8 × 8 × 8, 4 × 6 × 3, 7 × 7 × 2, 1 × 7 × 2, 2 × 2 × 4, 2 × 2 × 7, and 2 × 2 × 4 for orthorhombic ZrTe, hexagonal ZrTe, monoclinic ZrTe3, orthorhombic ZrTe5, orthorhombic Zr2Te, tetragonal Zr3Te, tetragonal Zr5Te4, and trigonal Zr5Te6, respectively. A total energy convergence of 5.0× 10−6 eV/atom was set during the structural optimization.
3. Results and discussion
3.1. Structural optimization
The crystal structures of Zr–Te compounds are shown in Fig.
 | Fig. 1. Crystal structures (Zr in blue, Te in orange) of (a) orthorhombic ZrTe, (b) hexagonal ZrTe, (c) monoclinic ZrTe3, (d) orthorhombic ZrTe5, (e) orthorhombic Zr2Te, (f) tetragonal Zr3Te, (g) tetragonal Zr5Te4, and (h) trigonal Zr5Te6. |
 | Fig. 2. Plots of density versus Zr content. |
3.2. Formation energy
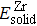
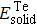
2 shows the calculated formation energy values of Zr–Te compounds. From our calculations, the Zr–Te compounds are found to be thermodynamically stable due to their formation energy values are all negative.
In order to evaluate thermodynamic stability, the formation energy values are calculated for these considered cases. The negative formation energy indicates that the reaction is an exothermic reaction and the final compound is thermodynamic stability. The computed equation of formation energy (Δ H) is given as follows:

| Table 2. Calculated formation energy values of Zr–Te compounds. . |
As shown in Fig.
 | Fig. 3. Plot of formation energy versus Zr content. |
3.3. Mechanical stability and properties3 . It is well known that the crystal is considered to be mechanically stable when the quadratic form from elastic constants’ matrix is always positive.[32] Hence, the Born stability criteria for hexagonal, tetragonal, trigonal, orthorhombic, and monoclinic structures are shown as follows:
The elastic constants of Zr–Te compounds are calculated to estimate their mechanical stability by the stress–strain calculations in their optimized crystal structures. The lattice energy for expanding elastic strain and the elastic constants are given as follows:[31]


for hexagonal phase,

for tetragonal phase,

for trigonal phase,

for orthorhombic phase,


In Table
| Table 3. Calculated elastic constants of Zr–Te compounds. . |
As is well known, the elastic constants are associated with the single crystal and the elastic modulus is linked with the polycrystal.[33–35] Then, we introduce three methods to calculate elastic modulus, i.e., the Voigt, Reuss, and Hill methods,[33–36] which are related to the upper, lower, and average bounds of elastic constants, respectively.
Tetragonal, orthorhombic, and monoclinic phases are







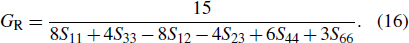
Then, bulk modulus B and shear modulus G are calculated by the Voigt–Reuss–Hill (VRH) method[33]









Table
| Table 4. Calculated values of bulk modulus B (in unit GPa), shear modulus G (in unit GPa), Young’s modulus E (in unit GPa), Poisson’s ratio v, sound velocity vm (in units m/s), Debye temperature ΘD (K), Vickers hardness Hv (GPa) and G/B ratio of Zr–Te compounds. . |
In Fig.
 | Fig. 4. Plots of bulk modulus B (GPa), shear modulus G (GPa), Young’s modulus E (GPa), Debye temperature ΘD (K), and sound velocity vm (m/s) of Zr–Te compounds versus atomic fraction Zr. |
| Table 5. Pearson correlation (Corr) and significant level (sig) of calculated bulk modulus B (GPa), shear modulus G (GPa), Young’s modulus E (GPa), Debye temperature ΘD (K), and sound velocity vm (m/s) for Zr–Te compounds. . |
3.4. Electronic properties
In order to show the electronic properties of Zr–Te compounds, the total densities of states (TDOSs) of Zr–Te compounds are shown in Fig.
 | Fig. 5. Plots of TDOS versus energy of ZrTe compounds. |
 | Fig. 6. Band structure of ZrTe5. |
The charge densities of Zr–Te compounds are calculated to further analyze the electronic states. In Fig. 
 | Fig. 7. Calculated charge densities of Zr–Te compounds: (a) ZrTe (Pnma) (010), (b) ZrTe (P-6m2) (111), (c) ZrTe3 (010), (d) ZrTe5 ( |
4. Conclusions
The first-principles calculations are carried out to investigate the geometrical structures, phase stability, mechanical properties and electronic structures of Zr–Te compounds. The negative formation energy values reveal that these compounds are thermodynamically stable. These compounds are also mechanically stable according to the mechanical stability criteria. The mechanical properties of Zr–Te compounds are calculated and discussed. The ZrTe5 has the lowest bulk modulus of 25.38 GPa, and Zr2Te has the largest bulk modulus of 119.93 GPa. For shear modulus and Young’s modulus, the biggest values both appear in ZrTe (P-6m2) phase, and the least values both appear in ZtTe5 phase. Correspondingly, the ZrTe (P-6m2) phase shows a maximum sound velocity, Debye temperature, and Vickers hardness, and the ZtTe5 phase presents the smallest value, which is due to the bonding force between atoms. The ZrTe possesses Pnma space group, and ZrTe3, Zr2Te, and Zr3Te belong to ductile compounds according to the analysis of G/B, and the others belong to brittle compounds. Moreover, the bulk modulus B, shear modulus G, Young’s modulus E, Debye temperature ΘD, and sound velocity vm are positive correlation with the increase of fraction Zr. The calculated electronic properties show that Zr–Te compounds have metallic characters except ZrTe5. Whether ZrTe5 is a metal or a semimetal needs further studying. The charge densities of Zr–Te compounds indicate the forming of σ bond between atom Zr and atom Te.
Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] | |
| [33] | |
| [34] | |
| [35] | |
| [36] | |
| [37] | |
| [38] | |
| [39] | |
| [40] | |
| [41] |