




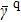

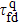
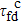
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Quantum intelligence on protein folding pathways
Cite this Article
Mao Wen-Wen, Lv Li-Hua, Ji Yong-Yun, Li You-Quan. Quantum intelligence on protein folding pathways. Chinese Physics B, 2020, 29(1): 018702 
Permissions
Quantum intelligence on protein folding pathways
† Corresponding author. E-mail:
Project supported by the National Key Research and Development Program of China (Grant No. 2017YFA0304304) and the National Natural Science Foundation of China (Grant No. 11935012).
Abstract
We study the protein folding problem on the base of our quantum approach by considering the model of protein chain with nine amino-acid residues. We introduce the concept of distance space and its projections on a XY-plane, and two characteristic quantities, one is called compactness of protein structure and another is called probability ratio involving shortest path. The concept of shortest path enables us to reduce the 388 × 388 density matrix to a 2 × 2 one from which the von Neumann entropy reflecting certain quantum coherence feature is naturally defined. We observe the time evolution of average distance and compactness solved from the classical random walk and quantum walk, we also compare the features of the time-dependence of Shannon entropy and von Neumann entropy. All the results not only reveal the fast quantum folding time but also unveil the existence of quantum intelligence hidden behind in choosing protein folding pathways.
1. Introduction
Protein folding problem has been an important topic in interdisciplinary field involving molecular biology, computer science, polymer physics as well as theoretical physics, etc. Levinthal noted early in 1967 that a much larger folding time is inevitable if proteins are folded by sequentially sampling of all possible conformations. It was widely assumed that a random conformational search does not occur in the folding process, for which various hypotheses with the help of a series of meta-stable intermediate states have been often proposed. There have been substantial theoretical models which are useful for understanding the essentials of the complex self-assembly reaction of protein folding with different simplifying assumptions, such as Ising-like model,[1,2] foldon-dependent protein folding model,[3] diffusion–collision model,[4,5] and nucleation-condensation mechanism.[6,7] However till now, this often brings in certain difficulties in connecting analytical theory to experimental results because some hypotheses can not be easily put into a practical experimental measurement since they often rely on various hypotheses.[8–12] As the approach of computing simulations introduced less hypotheses in comparison to those theoretical models, the atomistic simulations[13–15] have been also used to investigate the protein folding along with nowadays’ advances in computer science. All-atom computational method,[16] including physics-based and knowledge-based approaches, have provided useful insights on protein folding and design by building high-accuracy atomistic models of proteins. However, all those models are computationally costly for high-throughput folding studies and still need certain artificial hypotheses.
Recently, we proposed a quantum strategy[17] without making hypotheses to formulate protein folding as a quantum walk on a definite graph, in which we merely studied the model with six amino-acids as toy model. We know the shortest peptide chain in nature contains more than twenty amino-acid residues. Toward a genuine understanding, it is obligatory to study more complicate case with more residues. Here we consider the model with nine amino-acid residues and obtain that the folding time via our quantum approach is much shorter than the one obtained via classical random walk. Moreover, as the number of amino acid residues increases, the set of protein structures and the corresponding connection graph become more complicate, which drives us to introduce the projection method and several characteristic quantities. After calculating and analyzing those quantities, we find some new features on the folding pathways in addition to the fast protein-folding time. Our quantum approach that employs and combines the concepts of quantum walk and mean first-passage time not only reveals fast protein-folding time but also unveils the existence of quantum intelligence on the protein folding pathways, which is expected to open a significant new area of research.
2. Model description
where the geometric center of the protein is located at
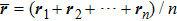
, and r k refers to coordinates of the k-th residues. Clearly, the compactness Ca represents the average distance from each residues to its geometric center of a given structure sa. We consider in this paper the case of n = 9 and obtain N9 = 388 through depth search algorithm (DSA). The 388 distinct structures that are unrelated by rotational, reflection or reverse-labeling symmetries[20] were plotted (see supplementary material, Table S1). The straight-line structure is labeled lastly as s388 for convenience since it is the farthest end of the depth-first algorithm search. We plot it together with the three most compact structures s186, s193, and s236 in Fig. 1 as an illustration. For the structure set
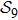
, the compactness of each structure is calculated (see supplementary material, Table S2). The largest value C = 2.222 refers to the completely unfolded straight-line structure s388, while the smallest value C = 1.073 refers to the most compact structure s186, s193, and s236.
In the course-grained model the protein is considered as a chain of non-own intersecting unit[18–21] (usually referring an amino acid residue) of a given length on the two-dimensional square lattice. We indicated recently[17] that for a protein with n amino-acid residues, there will be totally Nn distinct lattice conformations that distinguish various protein intermediate structures, which provide us a point set with Nn objects. We call such a point set as structure set and denote it by 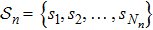
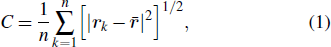 |
 | Fig. 1. Structure examples. There are 388 distinct structures for the amino-acid chain with 9 residues, thus the corresponding structure set 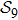 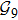 |
In terms of the lattice model, we introduced previously the concept of one-step folding to describe the protein folding process.[17] The one-step folding is defined by one displacement of an amino acid in one of the lattice sites: two protein structures are connected via one-step folding if their chain conformations differ in one site only. For example, in Fig. 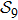
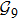
3. Distance space and its projections
where ρI, I = ∑aρaa, ρII, II = ∑bρbb,
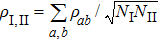
and
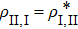
with a ∈ SI, b ∈ SII and NI, NII the total numbers of the objects in the corresponding subsets. Here the large density matrix stands for ρ(t) = |Ψ(t)〉〈Ψ(t)|.
If the aforementioned graph 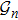
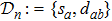
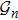
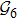
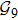
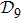
It will be very helpful to make a projection of the space 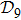
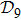
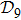
The color scatter diagram shown in Fig. 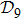
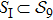
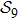

 | Fig. 2. Projection of the distance space  |
The density matrix ρ(t) describing quantum dynamics is of 388 × 388 that is too large to manifest some useful information. The above projection picture helps us to properly reduce the large density matrix to a 2 × 2 one to extract certain useful information, namely,
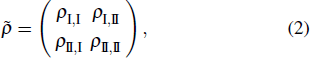 |
4. On the time evolution
in which
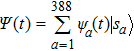
, and the expansion coefficients ψa(t) can be solved numerically at least. In our numerical calculation, we set ℏ and J to be unity and take the time step as Δt = 0.02. For the initial condition, |Ψ(0) = |s388〉, we solve the first-order differential equation (3 ) by means of Runge–Kutta method and obtain the magnitude of ψa(t) at any later time, t = j⋅Δt with j = 1,2,…
where Kab = Tab – δab with Tab being the probability-transition matrix. In the conventional classical random walk, the probability-transition matrix is determined by the adjacency matrix of an undirected graph, namely, Tab = Jab/deg(b) where deg(b) = ∑cJcb represents the degree of vertex-b in the graph.
Letting |sa〉 denote the state of a protein structure in the shape of the a-th lattice conformation, we will have a quantum Hamiltonian in a 388-dimensional Hilbert space 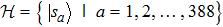

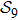
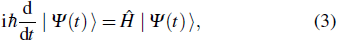 |
In order to compare with the protein folding problem in the classical literature, i.e., a random conformation search process, let us revisit the classical random walk. The continuous-time classical random walk[24] on a graph 
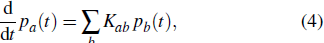 |
It is widely believed that the native structure of a protein possesses the lowest free energy.[26] This can be interpreted by the hydrophobic force that drives the protein to fold into a compact structure with as many hydrophobic residues inside as possible.[19] How fast does a protein initially in a straight-line structure fold into the most compact structure and what is the main behavior during the folding process will be important issues to study.
5. Folding time and folding pathway
where
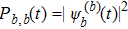
arises from the solution of Eq. (3 ) from another initial condition |Ψ(0)〉 = |sb〉. In the classical case, Pa,b and Pb,b refer to the pb(t) solved from Eq. (4 ), respectively, with initial conditions pc(0) = δac and pc(0) = δbc.
where τ0 represents the time period when the first-passage probability vanishes Fa,b(τ0) = 0 firstly.[17] Here a = 388 and c = 186 or 193. We know in a previous work that the quantum folding is faster than the classical folding with about four to six times even for the simplest model of 4 residues and it is faster than the classical folding with almost ten to hundred times or more for n = 6.[17] Here we calculate the folding time for n = 9 that is given in Table 1 . We can see that the quantum folding time
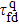
is much shorter than the classical folding time
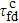
, and their difference becomes more significant in the case of n = 9 in comparison to that of n = 6.
where Γσ(t) = ∑b ∈ SσPa,b(t) with σ = I, II. Our result is plotted in Fig. 3 , we can see that the magnitude of γ changes from 0 to 1 monotonously during a short period of time for both quantum and classical cases. This implies that the probability on the shortest path is more favorable after leaving the initial state for it is on the denominator of γ. In order to rule out the ambiguity on the time scales about classical and quantum literature, we evaluate a mean ratio,
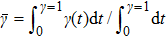
and show them in Table 1 . The mean ratio in quantum case
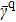
is smaller than that in classical case
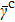
. This implies that the probability distribution is more aggregated on the shortest path in quantum case.
We let Pa,b(t) denote the probability of a state being the basis state |b〉 at time t if starting from the state |a〉 at initial time t = 0. Quantum mechanically, 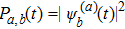
 |
As protein folding is the process that proteins achieve their native structures, the folding time is the case that the starting state is chosen as |s388〉 and the target states are the most compact states |sc〉. For example, they are |s186〉, |s193〉 for n = 9 as aforementioned. With the help of the first-passage probability solved from Eq. (
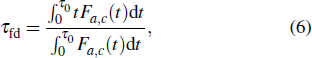 |
| Table 1. The probability ratio and the folding time. . |
In order to explore whether there are any intelligence hidden behind in choosing the protein-folding pathway, we compare the total probability on the shortest path and the other path by a probability ratio γ to demonstrate how it changes relatively after it leaves the initial state, namely,
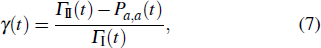 |
 | Fig. 3. The probability ratio γ. The time evolution of the folding process from s388 to s186 and s193 for (a) quantum walk and (b) classical random walk. |
6. More on folding behavior
Although the random walk reflects a stochastic process, it is characterized by the probability distribution defined on all the distinct structures that can always give us intuitive information if we calculate some weighted average of the entire system. We can attain the time dependence of mean distance away from any structure sa by calculating 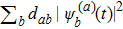
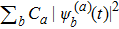
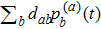
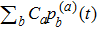
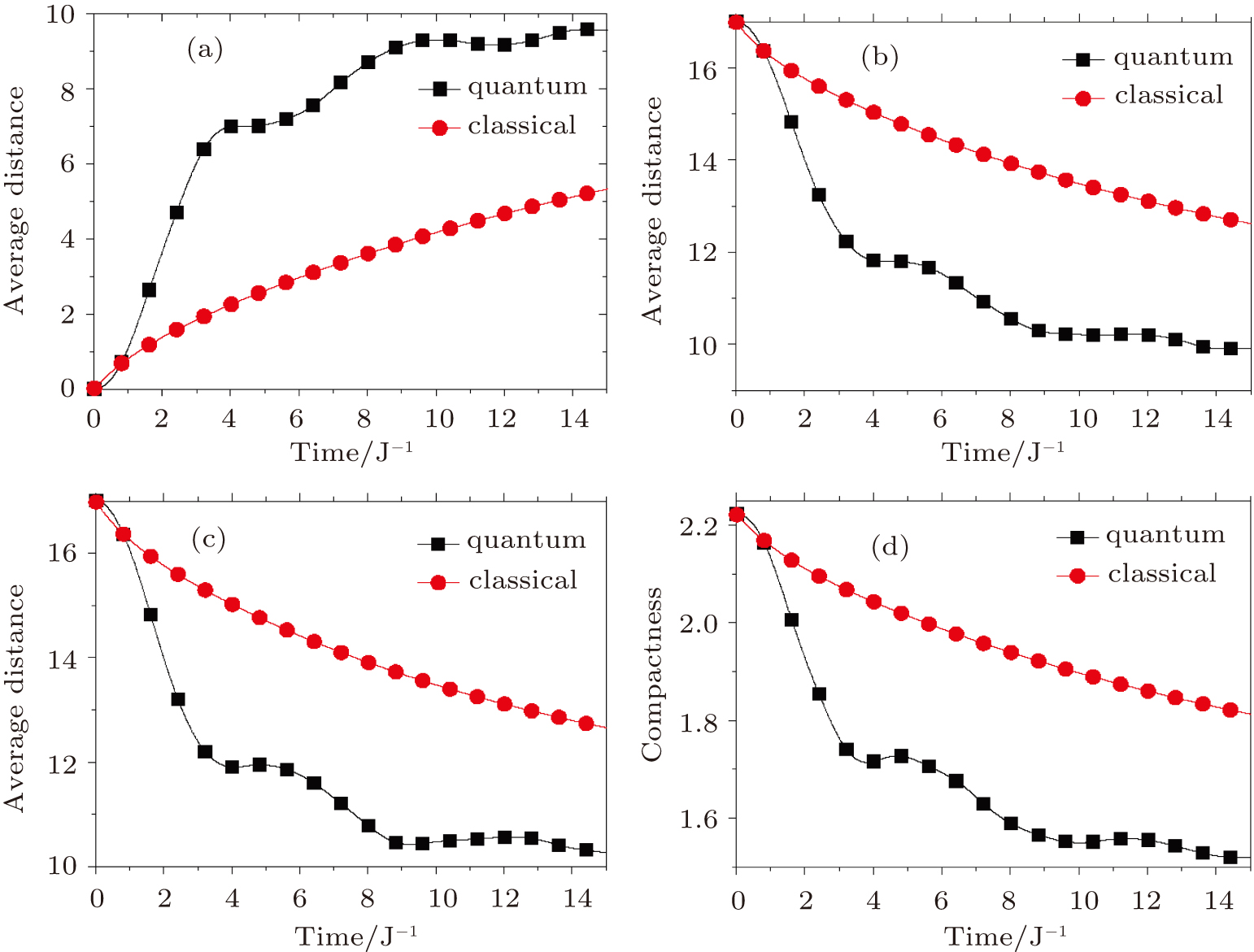 | Fig. 4. The time evolutions of average distance and compactness. (a) The average distance leaving away from the initial structure s388. (b) The average distance approaching to the most compact structure s186 while leaving from the structure s388. (c) The average distance approaching to the most compact structure s193 while leaving from the structure s388. (d) The time dependence of the overall average of compactness. |
As we know, quantum mechanically, the off-diagonal elements of a density matrix reflect certain quantum coherent properties, but our discussion till now have not yet involve it directly. The partition of entire structure set into subset by considering the concept of shortest path helps us to have a 2 × 2 density matrix. It is then worthwhile to calculate von Neumann entropy[32] 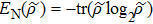
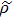

The time dependence of both von Neumann and Shannon entropies are plotted in Fig.
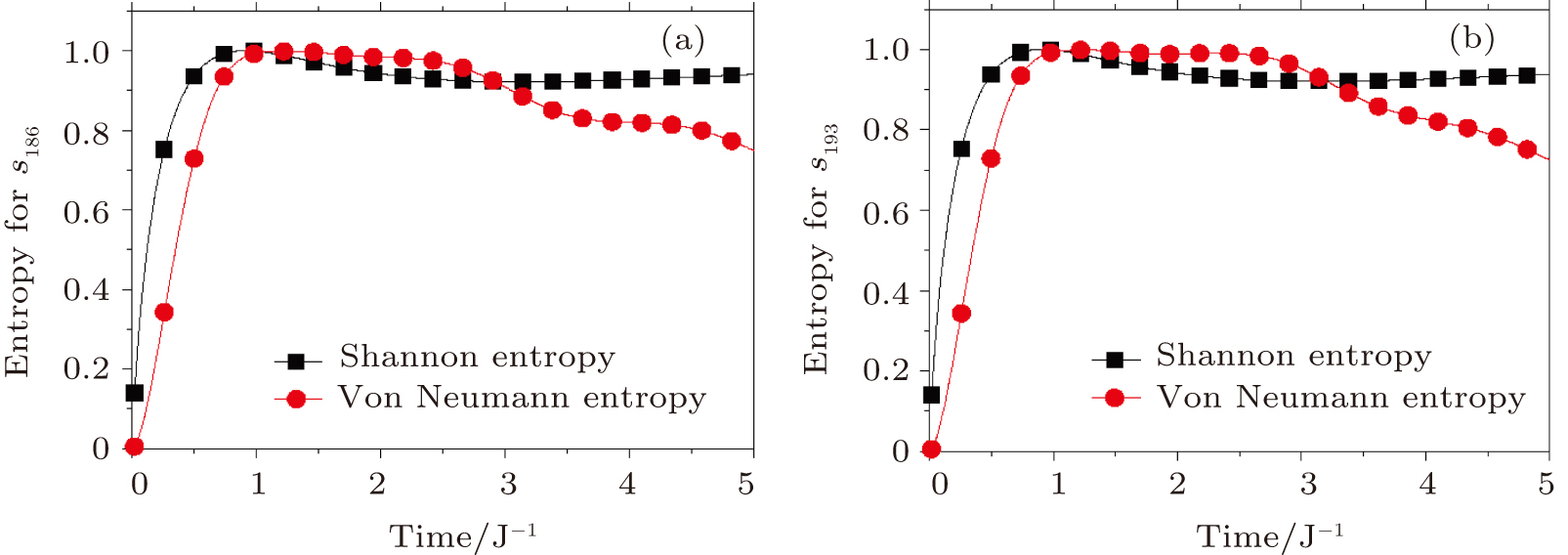 | Fig. 5. The time-dependence of the entropy. (a) The quantum and classical time evolution of the entropy from s388 to s186. (b) The quantum and classical time evolution of the entropy from s388 to s193. |
7. Summary
In the above, we studied the protein folding problem on the base of the quantum approach we proposed recently[17] by considering the model of protein chain with nine amino-acid residues. We have shown that the protein folding can be modelled as a quantum walk on the graph 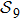
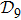
Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] | |
| [33] | |
| [34] | |
| [35] |