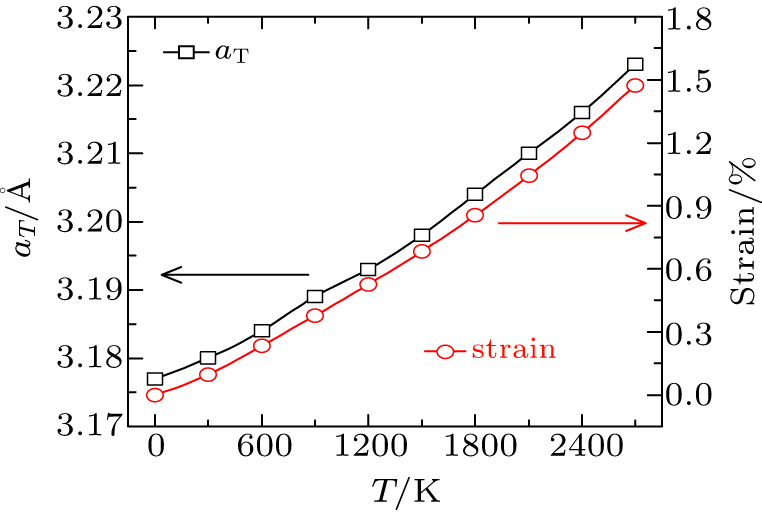
3.1. Verification of the calculationsThe calculated lattice constant of the tungsten at 0 K is 3.177 Å, which agrees well with the previous calculation results[29–31,33] and is slightly larger than the experimental value (3.165 Å[45]). Using Eq. (6), the relevant lattice constants (aT) at temperature T are calculated and presented in Fig. 1, which is used in the following calculation. Note that the lattice expansion corresponds to the isotropic tensile strain. Therefore, the corresponding lattice strain εaT are also presented in Fig. 1.
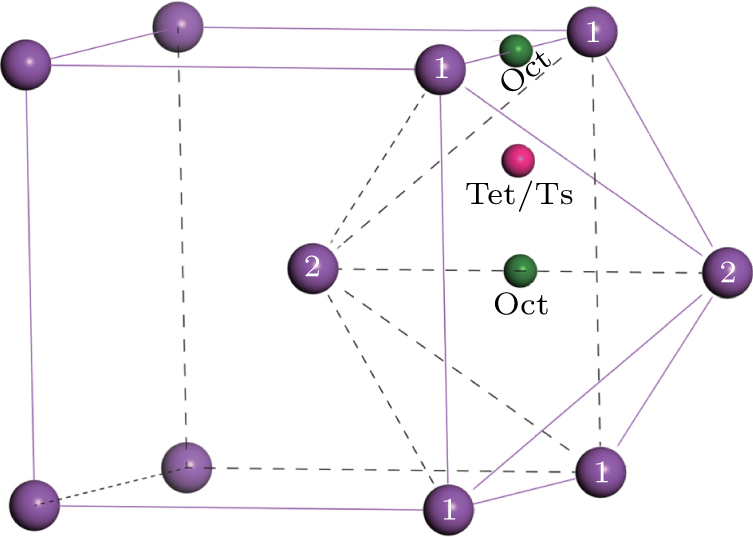
To verify the accuracy of our calculations, the solution and diffusion properties of interstitial carbon in tungsten at 0 K are calculated. Being small enough, carbon will occupy interstitial sites in bcc metal. Two possible interstitial sites, i.e. tetrahedral (Tet) and octahedral (Oct) sites, are considered. The formation energy is calculated by Ef = EWC – EW – EC, where EWC is the total energy of the tungsten lattice with a carbon atom at interstitial site, EW is the total energy of tungsten perfect supercell, and EC is the energy of carbon isolated. Using this equation, the formation energies of carbon in tetrahedral and octahedral sites are calculated to be −4.45 eV and −5.92 eV, respectively. This means that the carbon atom prefers to occupy octahedral interstitial site rather than tetrahedral interstitial site. For seeking the saddle point in the process of the carbon atom migration, the nudged elastic band (NEB) method is utilized. The interpolation points of the system are set 5 to construct a minimum energy path. According to our calculation, the minimum energy path for the interstitial carbon diffusion is found directly through a Tet, i.e., the path Oct→Tet→Oct shown in Fig. 2. Further calculations on the vibrational frequencies observed only one imaginary frequency at the state with a interstitial carbon atom at Tet. This confirms that the Tet is a saddle point for the interstitial carbon diffusion. Thus, the migration energy is simply given by the difference in energy between the states of interstitial carbon atom at Oct and Tet, i.e., 1.47 eV. The pre-exponential factor calculated by Eq. (5) is 2.54 × 10−7 m2/s. These calculated results are in good agreement with previously reported results (see Table 1).[31–33]
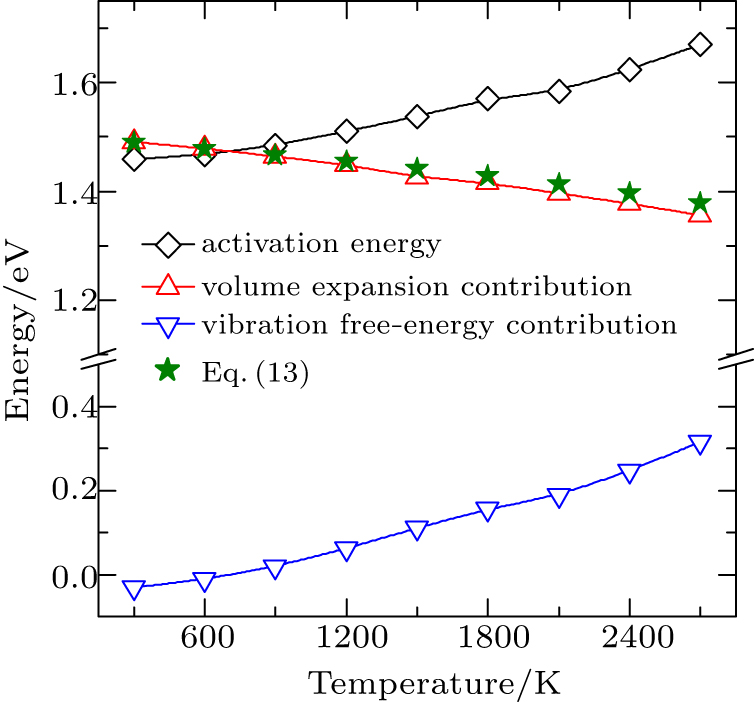
3.2. The temperature effects on the diffusionThe temperature effect on the activation energy can be evaluated by substituting Eq. (8) into Eq. (4), i.e.,
which can be divided into two parts, i.e., the contribution of the static energy
and the contribution of the vibration free energy
Figure
3 presents the calculated
Eact(
aT),
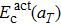
, and
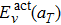
. It can be clearly seen that

decreases slowly and linearly with the temperature and its value decreases about 0.12 eV when the temperature increases from 300 K to 2700 K. On the contrary,
Eact(
aT) shows a positive dependence on the temperature, somewhat analogous to the behavior of
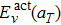
. This suggests that the major contribution to the increase of the diffusion activation energies across the temperature comes from the vibration free energy.
Table 2.
Table 2.
 Table 2. The carbon interstitial atom induced lattice stress (in GPa) in bcc tungsten. The compressive stress is negative and the tensile stress is positive. .
|
XX |
YY |
ZZ |
| σOct |
−0.977 |
−0.977 |
−2.638 |
| σTet |
−0.839 |
−2.155 |
−2.155 |
| Table 2. The carbon interstitial atom induced lattice stress (in GPa) in bcc tungsten. The compressive stress is negative and the tensile stress is positive. . |
Similar to the case of hydrogen in tungsten,[28,46] the dependence of
 on temperature results from the lattice expansion and can also be understand by the linear elasticity theory. According to linear elasticity theory, the solution energy of interstitial solute atom in system A under isotropic tensile strain ε can be expressed as
on temperature results from the lattice expansion and can also be understand by the linear elasticity theory. According to linear elasticity theory, the solution energy of interstitial solute atom in system A under isotropic tensile strain ε can be expressed as
Here
V is the volume of the supercell at equilibrium, and
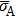
is the average stress induced by the interstitial solute.
ESolA is the solution energy of the solute in the system
A without strain, which can be calculated by
where
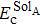
and
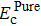
represent the static energy of the system
A with and without solute, and
ESoliso is the energy of the isolated solute atom. Thus, for interstitial carbon diffusion in tungsten,
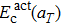
can be calculated by
according to Eqs. (
10), (
12), and (
13). Here
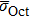
and
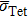
are the lattice stress induced by carbon located in Oct and Tet, respectively, whose values are summarized in Table
2. The calculated values of
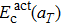
using Eq. (
14) are presented in Fig.
3, which are in close agreement with the DFT data. Therefore, the static energy contribution of the activation energy shows a linear-like negative dependence on the temperature.
Lattice expansion in general leads to a softening of vibrational modes, which in turn affects the vibration free energy and the partition functions (see section 2.1). In previous works on carbon diffusion in palladium[25] and hydrogen diffusion in tungsten,[28] researchers found that the softening of vibrational modes induced by lattice expansion has little effect on the temperature-dependent behavior of the vibration free energy. Similar results are also found in our present work. As shown in Fig. 4, the values of
 and ZTS/ZGS calculated using
and ZTS/ZGS calculated using
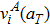 and
and
 , respectively, are almost identical. Thus, in the calculation of the interstitial solute diffusivity, one can use the vibration frequencies of the system with the a0 at all temperatures in order to save substantial amounts of computational time without significant loss of accuracy. Note that the explicitly values of
, respectively, are almost identical. Thus, in the calculation of the interstitial solute diffusivity, one can use the vibration frequencies of the system with the a0 at all temperatures in order to save substantial amounts of computational time without significant loss of accuracy. Note that the explicitly values of
 and ZTS/ZGS are still used in this work.
and ZTS/ZGS are still used in this work.
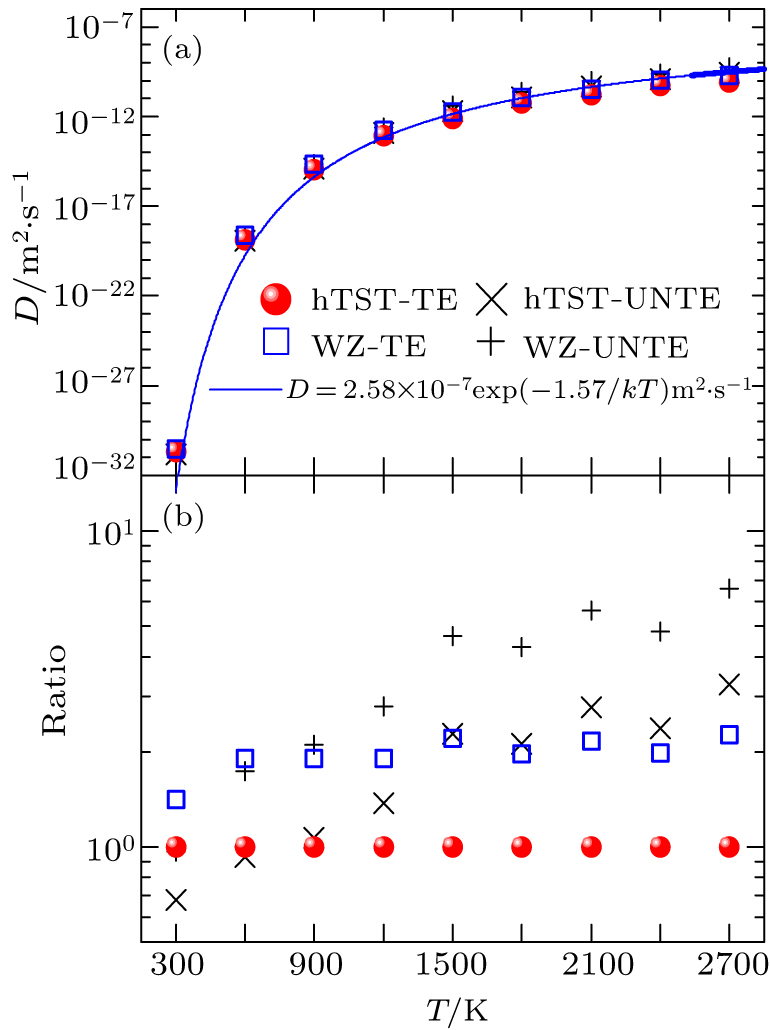
Using the two distinct methods of the calculation of the jump rate described in Section 2, the diffusion coefficients of carbon in tungsten with and without the temperature effect are calculated, i.e., the jump rate from the Wert–Zener model with and without the temperature effect (denoted by WZ-TE and WZ-UNTE, respectively) and the jump rate from the harmonic transition state theory of Eqs. (2) and (3) with and without the temperature effect (named by hTST-TE and hTST-UNTE, respectively). For the situation with the temperature effect, the above temperature-dependent lattice constant aT, activation energy Eact(aT) and vibration modes
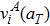 are used. When the temperature effect is excluded, we only use the diffusion activation energy (1.47 eV) and the vibration modes of the system with the DFT-optimized lattice parameter a0 = 3.177 Å. The calculated results of the carbon diffusion coefficients of these four cases are displayed in Fig. 5(a). It can be seen that the predicted diffusivity using these four different methods are quite close to each other. In order to more clearly evaluate their difference, the ratios of them to the case of hTST-TE are calculated and presented in Fig. 5(b). It can be seen that the calculated diffusivities with the WZ model are around twice as much as that with the hTST model. Meanwhile, both the WZ and hTST models would overestimate the diffusion coefficient at high temperatures when the temperature effect is ignored during the calculation. On the whole, the difference between them are less than an order of magnitude. This suggests that the temperature effect on the carbon diffusivity in tungsten is not significant. That is because the temperature has little effect on the pre-exponential factor (see Fig. 6). Meanwhile, although the increase in temperature leads to a change in activation energy, this change contributes less to the diffusion coefficient at high temperatures. Therefore, the predicted carbon diffusivity can be approximately fitted by the Arrhenius equation D = D0 exp(−Eact/kT) with D0 = 2.58 × 10−7 m2/s and Eact = 1.57 eV.
are used. When the temperature effect is excluded, we only use the diffusion activation energy (1.47 eV) and the vibration modes of the system with the DFT-optimized lattice parameter a0 = 3.177 Å. The calculated results of the carbon diffusion coefficients of these four cases are displayed in Fig. 5(a). It can be seen that the predicted diffusivity using these four different methods are quite close to each other. In order to more clearly evaluate their difference, the ratios of them to the case of hTST-TE are calculated and presented in Fig. 5(b). It can be seen that the calculated diffusivities with the WZ model are around twice as much as that with the hTST model. Meanwhile, both the WZ and hTST models would overestimate the diffusion coefficient at high temperatures when the temperature effect is ignored during the calculation. On the whole, the difference between them are less than an order of magnitude. This suggests that the temperature effect on the carbon diffusivity in tungsten is not significant. That is because the temperature has little effect on the pre-exponential factor (see Fig. 6). Meanwhile, although the increase in temperature leads to a change in activation energy, this change contributes less to the diffusion coefficient at high temperatures. Therefore, the predicted carbon diffusivity can be approximately fitted by the Arrhenius equation D = D0 exp(−Eact/kT) with D0 = 2.58 × 10−7 m2/s and Eact = 1.57 eV.
3.3. Comparison with experimentsThe agreement with experimental results is an extremely important criterion to verify theoretical predictions. For the interstitial diffusion of light elements in metals, the diffusivity data obtained at high temperatures are usually believed to be more reliable since they are likely less influenced by both surface and trapping effects. However, it is difficult to experimentally determine the temperature range without trapping effects due to the lack of the benchmarks. A comparison of experimental and theoretical data may hopefully solve this problem. Figures 6(a) and 6(b) exhibit the available experimental data of the carbon diffusivity in tungsten collated from the literature together with our calculated results here. Particularly, the pre-exponential factors and activation energies obtained by experimental measurement (Table 1) and first-principles calculation are also plotted in Figs. 6(c) and 6(d) to make a comparison and clearly display their difference. It can be seen from Figs. 6(a) and 6(b) that our predicted diffusion coefficients are in very good agreement with the experimental data when the temperature is above 1800 K. Moreover, our predicted activation energies are in the range of 1.58–1.67 eV at temperatures 1800–2700 K and increase with the temperature, which are comparable to those obtained by fitting the experimental data of carbon diffusion coefficients at high temperature (1.64 eV and 1.76 eV in the temperature ranges 1773–2073 K and 2073–3073 K, respectively, see Fig. 6(c). Unlike activation energy, our predicted diffusion pre-factors show no significant change with the temperature, which are around 1.3 × 10−7 and 2.5 × 10−7 for the models of hTST and WZ, respectively. They are roughly equivalent to most of the experimental results (see Fig. 6(d)). These consistencies not only indicate the reliability of our theoretical predictions but also shed some light on the experimental data at temperatures above 1800 K, less affected by trapping effects. When the temperature below ∼1800 K, our predicted diffusivity is larger than the experimental values. The difference between the experimental measurements and the theoretical predictions becomes larger as the temperature decreases. Moreover, as shown in Fig. 6(c), experiments give activation energies of 2.09 eV at 1523–1723 K, 2.32 eV at 1473–1873 K, and 2.56 eV at 1073–1143 K, which are obviously larger than our theoretical predictions. This indicates that the diffusion of carbon in the temperature range of T < 1800 K could be significantly affected by trapping effects. The defect-trapping effects on carbon diffusivity are very complex and related works are in progress.









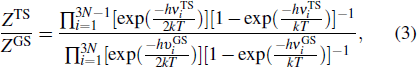


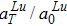


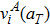

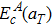




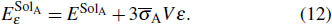

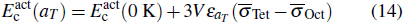



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}