























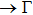

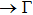








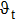
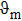
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Physical properties of ternary thallium chalcogenes Tl2MQ3 (M = Zr, Hf; Q = S, Se, Te) via ab-initio calculations
Cite this Article
Ateser Engin, Okvuran Oguzhan, Oztekin Ciftci Yasemin, Ozisik Haci, Deligoz Engin. Physical properties of ternary thallium chalcogenes Tl2MQ3 (M = Zr, Hf; Q = S, Se, Te) via ab-initio calculations. Chinese Physics B, 2019, 28(10): 106301 
Permissions
Physical properties of ternary thallium chalcogenes Tl2MQ3 (M = Zr, Hf; Q = S, Se, Te) via ab-initio calculations
† Corresponding author. E-mail:
Abstract
We have reported a first principles study of structural, mechanical, electronic, and thermoelectric properties of the monoclinic ternary thallium chalcogenes Tl2MQ3 (M=Zr, Hf; Q=S, Se, Te). The electronic band structure calculations confirm that all compounds exhibit semiconductor character. Especially, Tl2ZrTe3 and Tl2HfTe3 can be good candidates for thermoelectric materials, having narrow band gaps of 0.169 eV and 0.21 eV, respectively. All of the compounds are soft and brittle according to the second-order elastic constant calculations. Low Debye temperatures also support the softness. We have obtained the transport properties of the compounds by using rigid band and constant relaxation time approximations in the context of Boltzmann transport theory. The results show that the compounds could be considered for room temperature thermoelectric applications (
Keyword:electronic band structure;thermoelectric properties;figure of merit;ternary thallium chalcogens
1. Introduction
Recent studies about chalcogens have a variety of applications such as energy, magnetism, superconductivity, electronics, optoelectronics, and catalysis.[1–8] One of the popular topics within these is thermoelectricity, generating electricity by converting the temperature difference to an electrical voltage. For energy management in this way, the thermoelectric materials (TEM) become indispensable. As it is known that, the accomplishment of TEM addresses to dimensionless figure of merit (ZT) which is directly proportional to the power factor (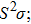

Especially, thallium containing chalcogenides are candidates of narrow band gap semiconductors because of their high atomic weight and so complex structure. Also, the high electronegativity of Tl compared to that of the other alkali metal ions is important for the band gap. Furthermore, it is also important that the presence of a lone pair in Tl+ ions causes anharmonicity in the lattice which has a remarkable effect on lowering the lattice thermal conductivity.[22–29] Tl2Ag12Se7, a new pnp conduction switching semiconductor (Eg = 0.8 eV) has just been synthesized, possessing a new structure type, namely, 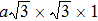
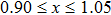

2. Method of calculation
All calculations have been accomplished by using the Vienna ab initio simulation package (VASP)[30–33] based on the density functional theory (DFT). The electron–ion interaction was considered in the form of the projector-augmented-wave (PAW) method with plane waves up to an energy of 450 eV,[33,34] an adequate value for studying the structural, elastic, and lattice dynamical properties. Perdew–Burke–Ernzerhof (PBE) type functional[35] within the generalized gradient approximation (GGA) has been used for the exchange and correlation terms in the electron–electron interaction. 17×17×13 Γ-centered Monkhorst–Pack[36] grid of k-points has been used for these monoclinic compounds.
To obtain the transport properties of Tl2MQ3 (M=Zr, Hf; Q = S, Se, Te) compounds, we have used the rigid-band approximation and semiclassical Boltzmann theory using BoltzTraP code.[37] For the calculations of transport coefficients (electrical conductivity σ, thermopower S, and electronic contribution of thermal conductivity ke), we need to calculate the velocities as a function of k for different bands. To get accurate electronic velocities, it is necessary to have a dense k-points mesh.[38] For this reason, we used 34×34 ×26 Monkhorst–Pack k-points sampling to obtain the band structure.
3. Results and discussion
3.1. Structural properties
We assume that the compounds Tl2MQ3 (M = Zr, Hf; Q = S, Se, Te) crystalize in monoclinic structure, similar to Na2FeO3-type, with 
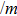
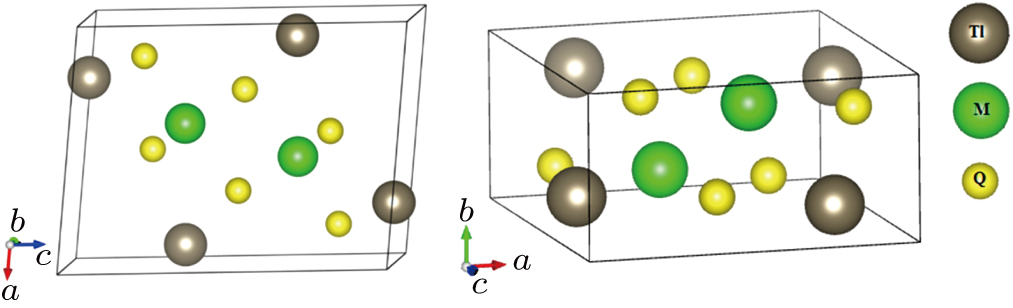 | Fig. 1. Unit cell arrangement of the Tl2MQ3 compounds with Na2FeO3-type structure. |
| Table 1.
The calculated atomic positions for Tl2MQ3 (M=Zr, Hf; Q=S, Se, Te) compounds (All of them are in 2e (x, 1/4, z) wyckoff positions). . |
We have fully optimized the lattice constants and ionic positions by taking the experimental values from Ref. [29]. The obtained equilibrium lattice parameters, volumes, and total energies are listed in Table
| Table 2.
Calculated equilibrium lattice parameters (a, b, and c in Å), the angle (β in degree), total energies (E0, eV/f.u.), formation enthalpy ( |
The thermal stability is calculated by using the formation enthalpy expression. Formation enthalpy 
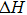

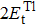
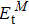
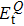


3.2. Electronic properties
The electronic band calculations have been performed for the titled compounds in monoclinic 

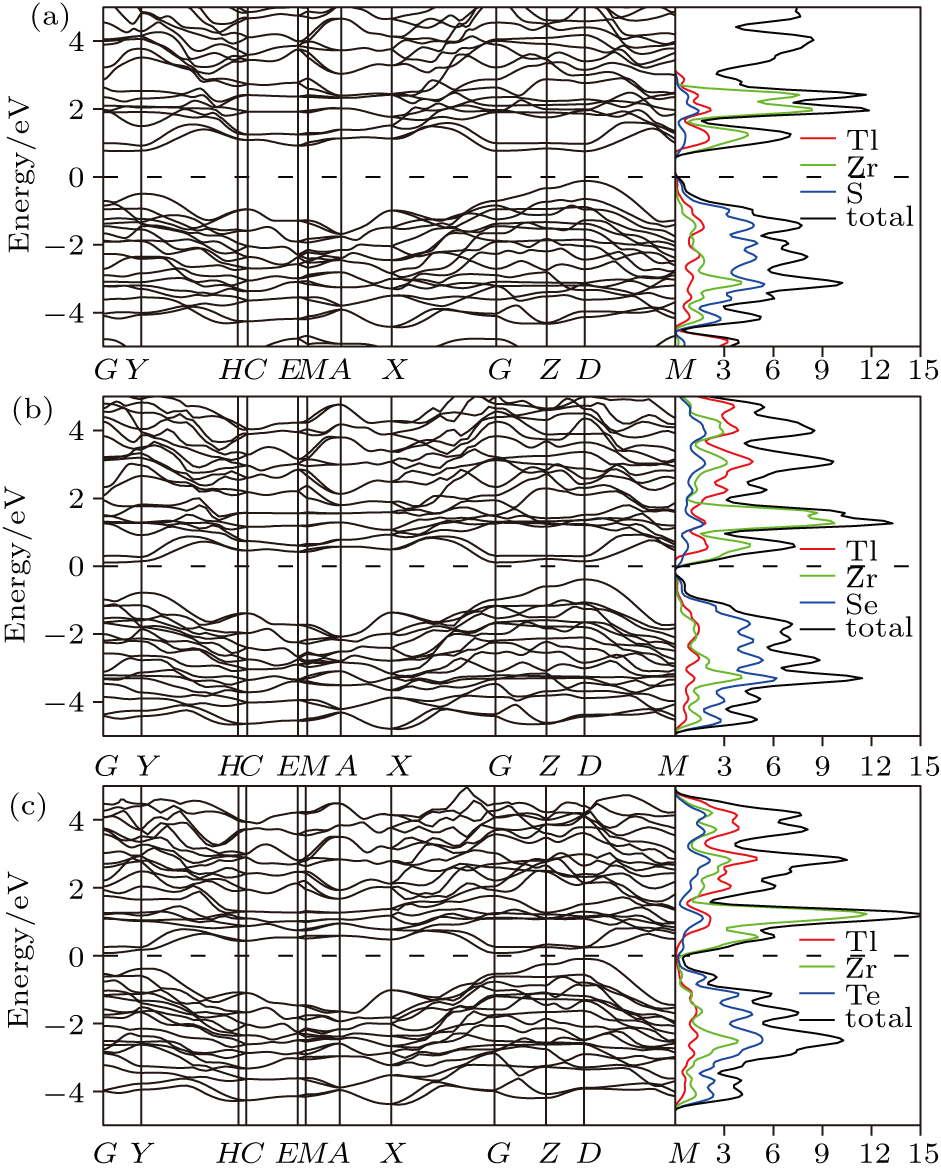 | Fig. 2. The electronic band structure and density of states for (a) Tl2ZrS3, (b) Tl2ZrSe3, and (c) Tl2ZrTe3. |
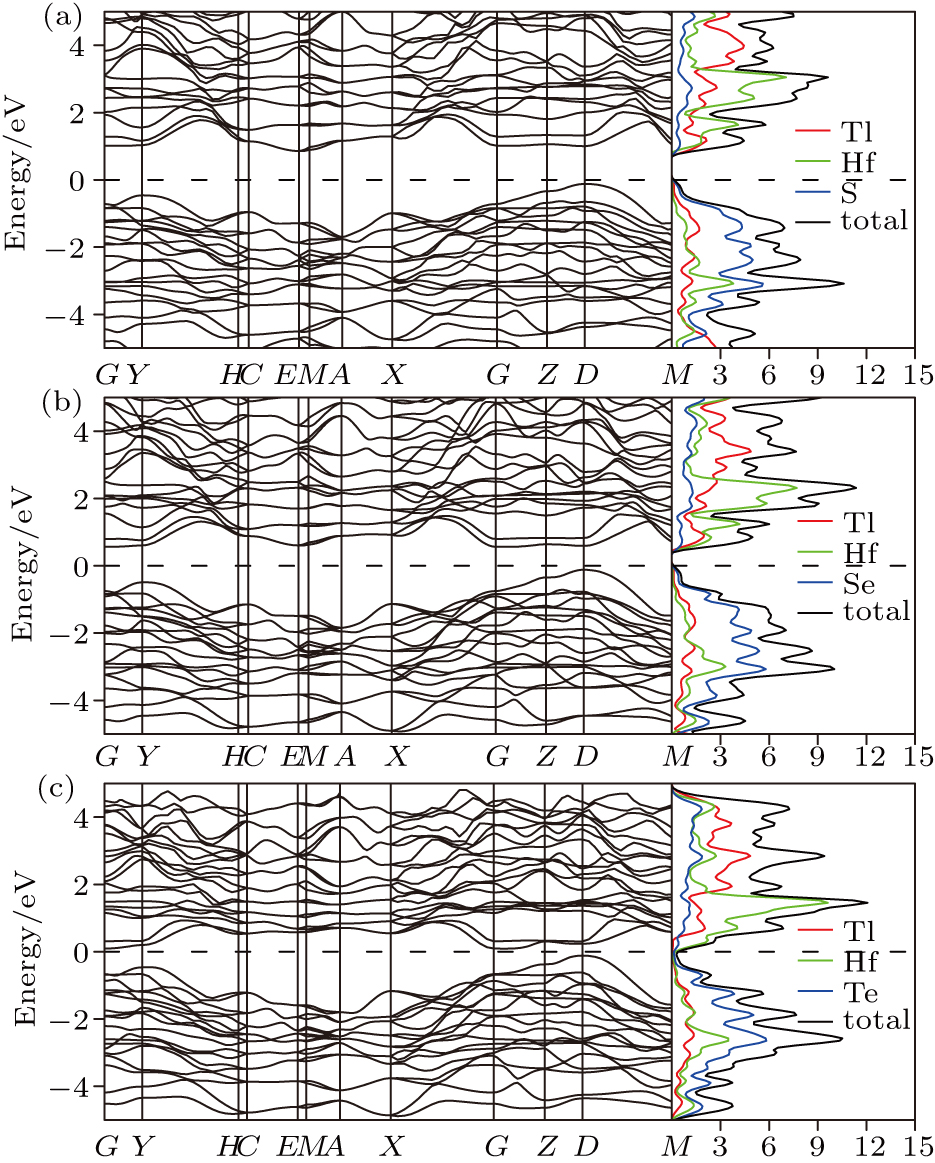 | Fig. 3. The electronic band structure and density of states for (a) Tl2HfS3, (b) Tl2HfSe3, and (c) Tl2HfTe3. |
As shown in Figs. 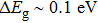
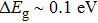
| Table 3.
The calculated band gaps and for Tl2ZrS3,Tl2ZrSe3, Tl2ZrTe3, Tl2HfS3, Tl2HfSe3, and Tl2HfTe3 compounds. . |
The total and projected densities of states of the studied compounds have been calculated and shown in Figs.
We have also considered the electronic charge densities of the related compounds for (
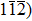
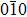

 | Fig. 4. The contour plot of charge density for (a) Tl2ZrS3, (b) Tl2ZrSe3, and (c) Tl2ZrTe3. |
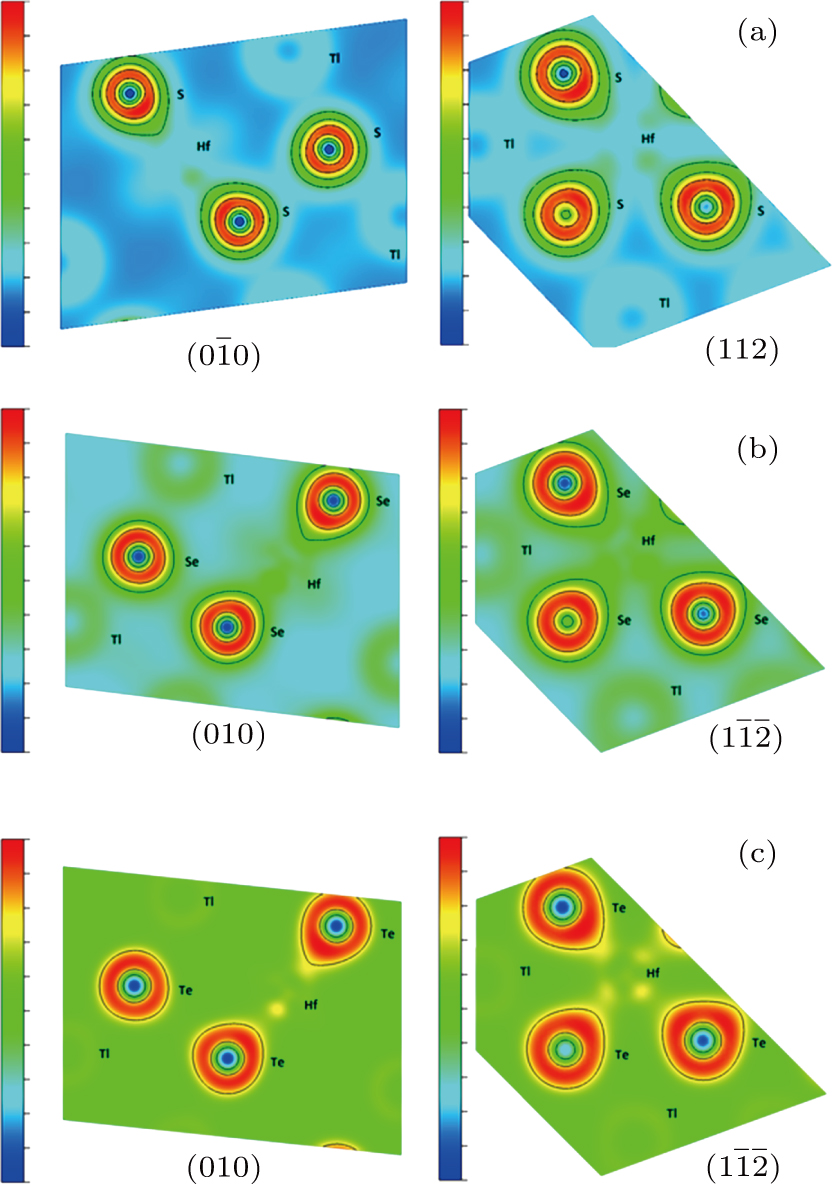 | Fig. 5. The contour plot of charge density for (a) Tl2HfS3, (b) Tl2HfSe3, and (c) Tl2HfTe3. |
3.3. Mechanical properties
3.3.1. Elastic properties
An elastic tensor provides the description of all the responses of a crystal to external forces within the elastic limits. It is one of the fundamental properties that explain the interatomic bonding in a system, and can be correlated with many mechanical, thermal properties for searching process of new materialʼs discovery and design.[35] We have computed the second order elastic constants (Cij) using the stress–strain methodology embedded in VASP.[30] The calculated Cij for the titled compounds shown in Table
| Table 4.
The calculated elastic constants (Cij, GPa) for Tl2MQ3 (M = Zr, Hf; Q = S, Se, Te). . |
Elastic constants C11, C22, and C33 measure the resistance to linear compression in the x-, y-, and z-directions, respectively. All compounds resist to compression in y-direction better than that in the other directions, so the compressibility from higher to lower is 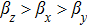
3.3.2. Polycrystalline properties
The bulk modulus B (a resistance to shape change under pressure) and shear modulus G (a resistance to deformations upon shear stress)[40] have been obtained from the Voigt–Reuss–Hill approximations[41] by using the calculated elastic constants. The Youngʼs modulus E and Poisson ratio ν are calculated through 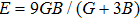
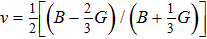
| Table 5.
Calculated bulk modulus B (GPa), isotropic shear modulus G (GPa), Youngʼs modulus E (GPa), Poissonʼs ratio ν, B/G ratio, Vicker hardness Hv, (GPa) Debye temperature |
Their shear moduli are almost similar to each other. However, since the large shear modulus is an indication of the more pronounced directional bonding between atoms.[42] It can generally be said that directional bonding of Tl2ZrS3 is more notable than that of the others due to its high value. On the other hand, a larger C44 elastic constant result with a larger shear modulus is not valid for our compounds.[43]
A greater Youngʼs modulus indicates a stiffer material, it is a measure of the resistance against uniaxial tensions.[44] Tl2ZrS3 has the largest bond strength among the considered systems, resulting in more resistivity to any deformation. A ductile material can be accessed with the help of Pughʼs ratio (B/G). Since all of the ratios are lower than 1.75, which is the critical starting value of ductility, all our considered systems show a brittle behavior.
The Poisson ratio is a measure of compressibility. when the Poisson ratio is close to 0.5, the material has tendency to be incompressible.[45] Here, the calculated Poisson ratio is about 0.23, which indicates that these materials are less compressible and stable against elastic deformation.[46] The value of Poisson ratio is also indicative of the degree of directionality of the covalent bonding. The Poisson ratio is about 0.1, 0.25, and 0.33 for covalent, ionic, and metallic materials, respectively.[47] Since the values are about 0.23, ionic contribution in the intra-atomic bonding is dominant with no central interatomic forces for the considered structures due to the lower limit of 0.25 for the central force solids. According to Frantsevich rule, the critical value of Poisson ratio of material is 0.33. Frantsevich et al. indicated that if the Poisson ratio is less than 0.33, the materials show brittle behavior.[48] This supports our results from the analysis of Pughʼs ratio (B/G).
The elastic and plastic properties of a material can be related to its hardness. The compounds are soft materials, since all of the calculated Vickers hardness values are lower than 10 GPa (see Table
As an important fundamental parameter, the Debye temperature (
3.3.3. Elastic anisotropy
Elastic anisotropy affects many physical processes such as plastic deformation development in crystals, enhancement of positively charged defect mobility, microscale cracking, phonons in crystallites, etc.[52] The degree of anisotropy in the bonding between atoms in different planes (A1, A2, and A3) could be measured by means of the shear anisotropic factors. Any value smaller or greater than 1, which is the isotropy criterion of crystals, is the indication of shear anisotropy. In addition, we have also calculated the percentage elastic anisotropy of bulk and shear moduli. In these expressions, the zero value correlates to the elastic isotropy and the value of 100% pinpoints the largest elastic anisotropy. Our results are given in Table
| Table 6.
The calculated shear anisotropy factors (A1, A2, and A3) and the percentage of anisotropy in the bulk and shear (AB and AG). . |
3.4. Transport properties
We have calculated the transport properties of the Tl2MQ3 compounds based on the Boltzmann transport equation under a rigid band and constant relaxation time approximations (RTA) which are very helpful in evaluating thermoelectric performance of materials based on theoretical band structure calculations,[53,54] i.e., the chemical potential shifts up or down from the intrinsic limit depending on the doping type and degree of doping and keeping the band structure unchanged.[55] In the rigid band approximation, the band structure is considered to be unaffected by doping, which only leads to change of the chemical potential. Boltzmann transport equations have a derivative of Fermi function with respect to the band structure and varies significantly within a small range near the chemical potential. So, in the calculations, instead of dependence of relaxation time on both the band index and the k vector direction, we have considered a direction independent τ, i.e., an almost constant relaxation time, which is a rational good approximation to express the transport coefficients.[56,57] Within RTA, the Seebeck coefficient can be calculated without any adjustable parameter.[58] However, the electronic conductivity has to be calculated with respect to the relaxation time and hence instead of calculating the absolute power factor (

Within the constant relaxation time approximation, the value of relaxation time is fixed to be in the order of 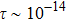
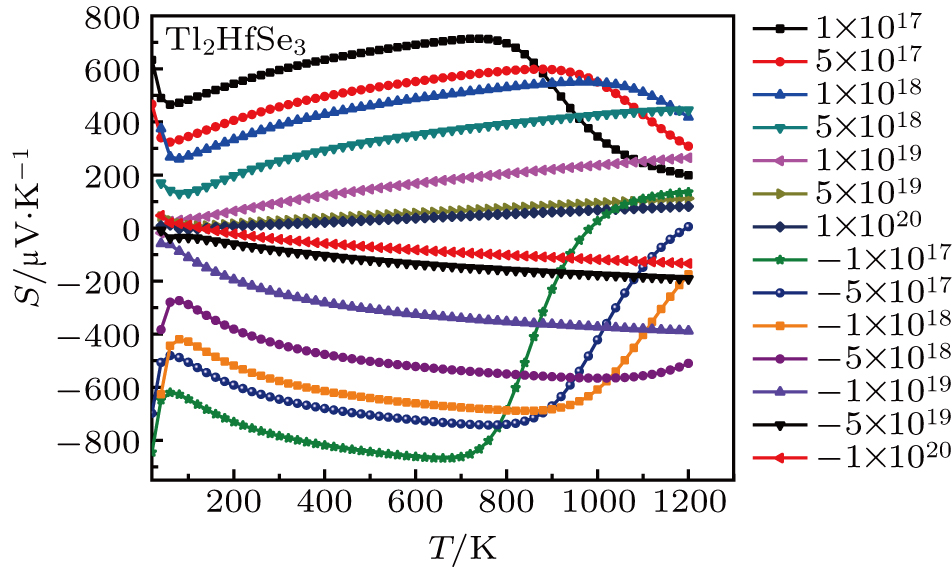 | Fig. 6. The Seebeck coefficient of Tl2HfSe3 at different concentration (cm−3) and temperature. |
The Seebeck coefficient of Tl2HfSe3 as a function of p-type and n-type carrier concentration at 300 K is shown in Fig.
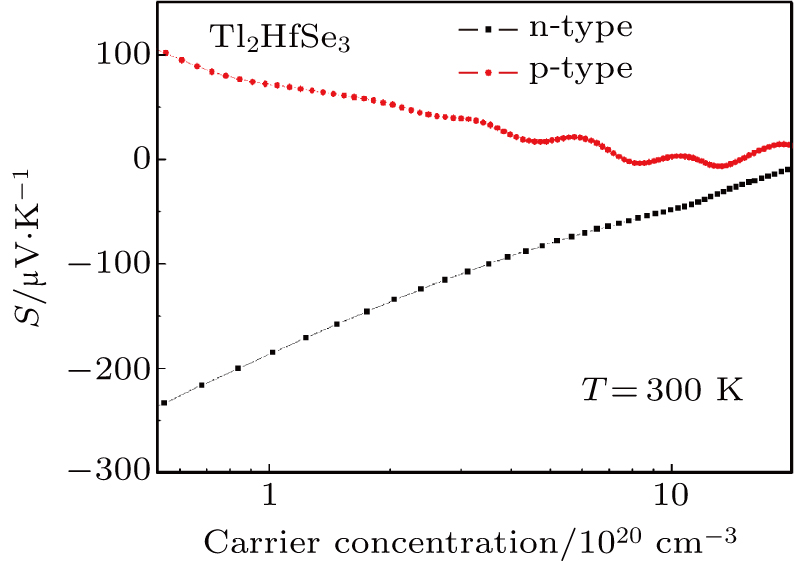 | Fig. 7. The Seebeck coefficient of p-type and n-type Tl2HfSe3 as a function of carrier concentration at 300 K. |
The calculated Seebeck coefficient (S), electrical conductivity (σ/τ), electronic thermal conductivity (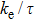



The Seebeck coefficient is an important tool, which provides a sensitive test of the electronic structures of the materials in close proximity to the Fermi energy. Large values of Seebeck coefficient are desired for efficient TE devices. The calculated S of the titled compound as a function of μ at different temperatures is illustrated in Fig. 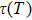
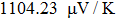
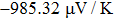

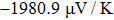
 | Fig. 8. The calculated Seebeck coefficient as a function of chemical potential at 300 K, 600 K, and 900 K for Tl2HfSe3. |
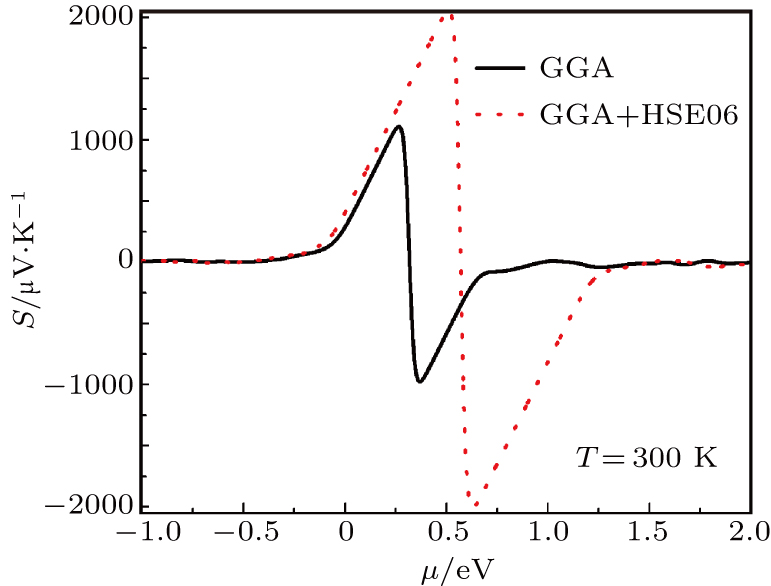 | Fig. 9. The calculated Seebeck coefficient as a function of chemical potential at 300 K for Tl2HfSe3. |
Electrical conductivity σ in units of τ has been calculated as a function of the chemical potential μ at three temperatures (300 K, 600 K, and 900 K) given in Fig. 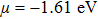
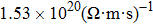
 | Fig. 10. The calculated electrical conductivity (in units of relaxation time τ) as a function of the chemical potential at 300 K, 600 K, and 900 K for Tl2HfSe3. |
While lattice vibrations are mostly subject to the heat conduction in semiconductors, free electrons are commonly the key of thermal conductivity in metals.[62] The thermal conductivity involves two parts as 

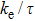
 | Fig. 11. Electronic thermal conductivity ke as a function of chemical potential at 300 K, 600 K, and 900 K for Tl2HfSe3 (the zero at low T corresponds to the undoped systems). |
It is important to increase the value of P as much possible as without increasing the total thermal conductivity k.[65] The P is given in Fig. 
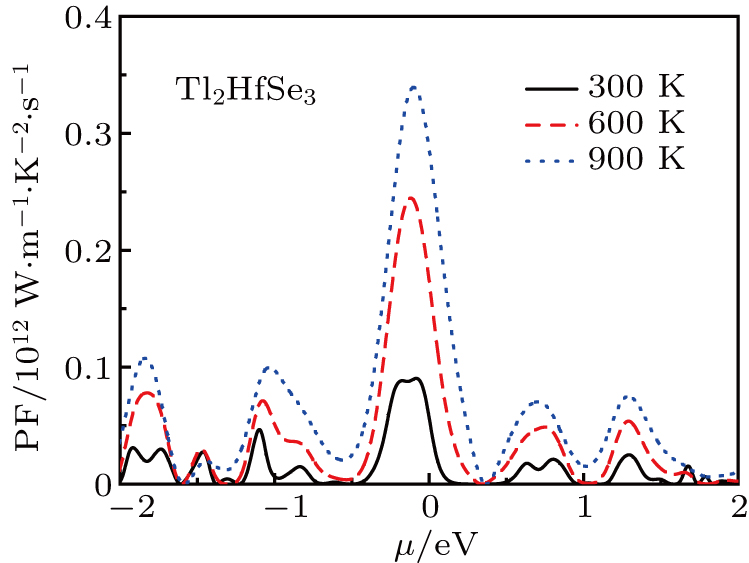 | Fig. 12. Power factor of Tl2HfSe3 at 300 K, 600 K, and 900 K as a function of the chemical potential. |
In the figure of merit calculations, we have only considered the electronic part of ZT because of not know anything (theoretically) about the lattice thermal conductivity. So, we define 



The 
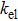
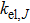
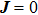

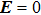


 | Fig. 13. Figure of merit 
|
Seebeck coefficient S, electrical conductivity 

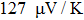
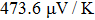



 | Fig. 14. The calculated (a) Seebeck coefficient, (b) electrical conductivity (in units of relaxation time τ) (c) thermal conductivity (in units of relaxation time τ), and (d) figure of merit 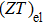
|
In order determine the thermoelectric figure of merit of Tl2HfSe3, the thermal conductivity including the kl of lattice and the ke of electron needs to be calculated. The lattice thermal conductivity kl equation derived by Slack[67] is given as

For Tl2HfSe3, at T = 300 K, using relaxation time 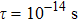



4. Conclusion
We have studied the structural, mechanical, electronic, and thermoelectrical properties of monoclinic Tl2MQ3 (M = Zr, Hf; Q = S, Se, Te) compounds by performing first-principles calculations. The calculated lattice parameters are in agreement with the available experimental data. The other original features of the present calculations involve the elastic, mechanical, and thermoelectrical results, which have not been considered previously. The analysis of the elastic constants indicates the mechanical stability and that the compounds have a brittle and soft nature with respect to the B/G (∼1.5), Poissonʼs ratio (∼0.23), and Hv (∼3 GPa) criteria. All studied systems are semiconductors in nature, which is also supported by the computed band structures, having an indirect band gap except for the Tl2ZrS3 compound. The total and projected densities of states of the studied compounds indicate that while Q (S, Se, and Te) elements are dominant in the conduction band, the leading elements of the valance band are M (Hf and Zr). The electronic transport properties are calculated as a function of the chemical potential and the highest value for 
Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] | |
| [33] | |
| [34] | |
| [35] | |
| [36] | |
| [37] | |
| [38] | |
| [39] | |
| [40] | |
| [41] | |
| [42] | |
| [43] | |
| [44] | |
| [45] | |
| [46] | |
| [47] | |
| [48] | |
| [49] | |
| [50] | |
| [51] | |
| [52] | |
| [53] | |
| [54] | |
| [55] | |
| [56] | |
| [57] | |
| [58] | |
| [59] | |
| [60] | |
| [61] | |
| [62] | |
| [63] | |
| [64] | |
| [65] | |
| [66] | |
| [67] | |
| [68] |