










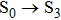

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Ab initio investigation of excited state dual hydrogen bonding interactions and proton transfer mechanism for novel oxazoline compound
Cite this Article
Wang Yu-Sheng, Jia Min, Zhang Qiao-Li, Song Xiao-Yan, Yang Da-Peng. Ab initio investigation of excited state dual hydrogen bonding interactions and proton transfer mechanism for novel oxazoline compound. Chinese Physics B, 2019, 28(10): 103105 
Permissions
Ab initio investigation of excited state dual hydrogen bonding interactions and proton transfer mechanism for novel oxazoline compound
† Corresponding author. E-mail:
Abstract
Owing to the importance of excited state dynamical relaxation, the excited state intramolecular proton transfer (ESIPT) mechanism for a novel compound containing dual hydrogen bond (abbreviated as “1-enol”) is studied in this work. Using density functional theory (DFT) and time-dependent density functional theory (TDDFT) method, the experimental electronic spectra can be reproduced for 1-enol compound. We first verify the formation of dual intramolecular hydrogen bonds, and then confirm that the dual hydrogen bond should be strengthened in the first excited state. The photo-excitation process is analyzed by using frontier molecular orbital (HOMO and LUMO) for 1-enol compound. The obvious intramolecular charge transfer (ICT) provides the driving force to effectively facilitate the ESIPT process in the S1 state. Exploration of the constructed S0-state and S1-state potential energy surface (PES) reveals that only the excited state intramolecular single proton transfer occurs for 1-enol system, which makes up for the deficiencies in previous experiment.
Keyword:excited state intramolecular proton transfer;potential energy surface;intramolecular charge transfer;infrared vibrational spectra
1. Introduction
As one of the most fundamental non-covalent weak interactions, hydrogen bond exists anywhere in our natural life. In the past few decades, more and more researches have been devoted to investigating the hydrogen bonding interactions.[1–3] In fact, hydrogen bond can be represented in the X–
It cannot be denied that most of chemical and biological systems exhibit multiple hydrogen bonding interactions. Therefore, the case about ESIPT process involved in single hydrogen bonding wire is not enough to imitate the biological behaviors. Therefore, to further explore the excited state behaviors along with multiple hydrogen bonds, one pays attention to the excited state single or double proton transfer reaction along with double hydrogen bonds since this kind of case should be the most fundamental way to further investigate multiple proton behavior in future.[29–35] For example, Krishnamoorthy et al. theoretically explored the double excited state intramolecular proton transfer behavior for the novel 3,5-bis(2-hydroxyphenyl)-1 H-1,2,4-triazole (bis-HPTA).[29] The new type of sequential proton transfer is referred to as proton transfer triggered proton transfer mechanism.[29] Tang et al. elaborated the intramolecular proton relay behaviors and confirm the undergoing ESDPT reaction process.[30] Song et al. compared the intermolecular dual hydrogen bonding effects for 3-hydroxyisoquinoline dimer and acidity compound.[31] They clearly clarified the detailed intermolecular dual PT behaviors. In addition, as a classical chemical system, 7-azaindole dimer has been explored by Crepo-Otero et al. in detail.[32] According to their simulated results, Crepo-Otero et al. presented that the stepwise mechanism is not consistent with the topography of the excited state.[32] As a whole, the investigation about ESPT reaction along with multiple hydrogen bonds is very important,[33–35] which plays a role in helping researchers to gain a more in-depth understanding of multiple proton behaviors in excited state.
Very recently, Reis et al. designed and synthesized a novel intramolecular dual hydrogen bond compound dimethyl-


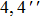
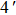
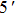
In this present work, therefore, we mainly pay attention to the excited state dynamic behavior for 1-enol compound by using theoretical simulation manner. We show the corresponding structures for 1-enol compound in Fig.
 | Fig. 1. Relative structures for 1-enol, 1-spt, 1-dpt, and their non-hydrogen bonding 1-open forms, where dual intramolecular hydrogen bonds are referred to as O1–

|
2. Theoretical methods
In this work, all calculations about structures and the stable energy for 1-enol system and its tautomers have been performed by using Gaussian 09 program.[50] And according to the DFT and TDDFT method with the Becke’s three-parameter hybrid exchange function combined with the Lee–Yang–Parr gradient-corrected correlation functional (B3LYP) as well as the triple-ζ valence quality with one set of polarization functions (TZVP) basis set,[51–54] we perform the quantum chemical simulations. To consider the solvent effect (DMSO) adopted in previous experiment,[36] in this work, we select the integral equation formalism version of the polarizable continuum model (IEFPCM).[55,56] The ground state structures for all the structures involved in this work are optimized by using the DFT method, and the vibrational frequencies at the optimized structures are calculated by using the same DFT method to verify that the optimized configurations correspond to the local minima on the S0-state potential energy surface (PES). Then, the photo-excitation process and corresponding charge redistribution are calculated by using the TDDFT method based on the optimized S0-state 1-enol structure. The first singlet excited state of each tautomers is relaxed by using the TDDFT method to obtain its minimum energy geometry. The frequency calculations of selected excited state minima are also calculated to verify that they correspond to the local minima on the S1-state PES. No constraints are imposed on symmetry, bond length, bond angle and dihedral angle in the simulation of geometry optimization. The PES of S0 state and the PES of S1 state are constructed each as a function of hydroxide radical length in a range from 0.9 Å to 2.1 Å in steps of 0.1 Å based on DFT and TDDFT method coupling with B3LYP/TZVP level in DMSO solvent, respectively.
The self-consistent field (SCF) convergence threshold of energy for each of S0- and S1-state optimizations is set to be 10−8 (default setting is 10−6). The harmonic vibrational frequencies of S0 and S1 states are determined by the diagonalization of Hessian matrix. Excited-state Hessian matrix can be obtained by numerical differentiation of the analytical gradients via central differences and default displacements of 0.02 Bohr. And the infrared intensities are determined by the gradients of the dipole moment.
3. Results and discussion
The 1-enol possesses dual intramolecular hydrogen bonds and a symmetrical structure. Therefore, we can locate three kinds of structures (i.e., 1-enol itself, its single proton-transfer 1-spt form, and its double-proton transfer 1-dpt form) in S0 and S1 states (Fig. 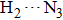


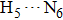
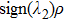

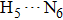
 | Fig. 2. Total electron density isosurface map with molecular electronic potential (MEP) for 1-enol structure, where values are selected from negative (red) to positive (blue): −0.05 a.u.–0.05 a.u. |
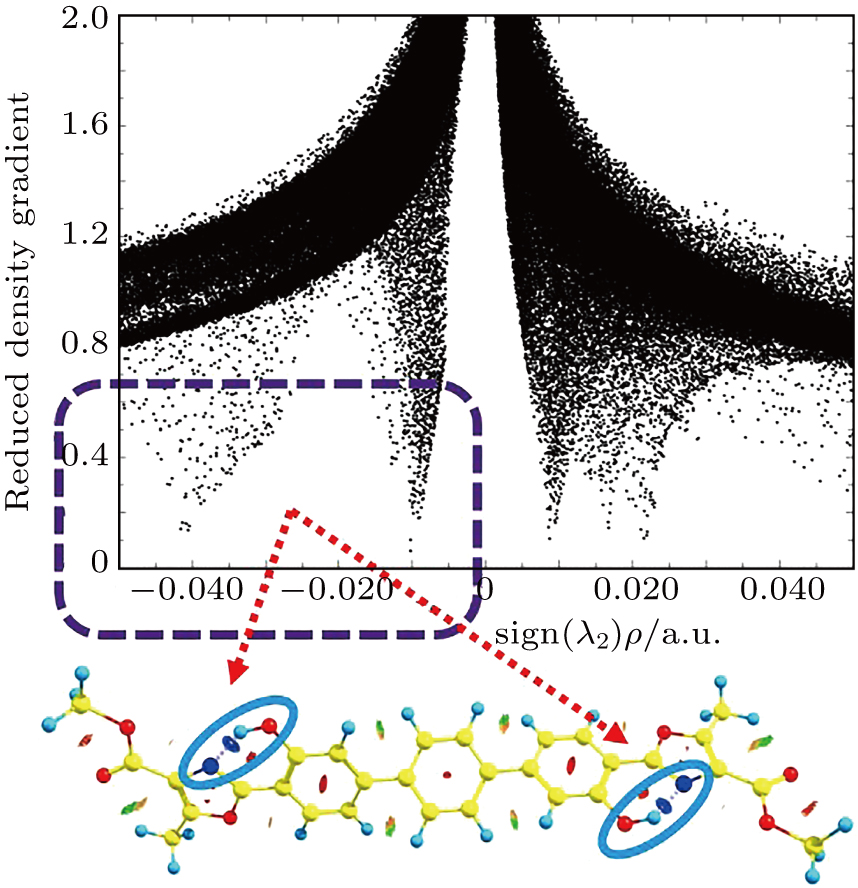 | Fig. 3. RDG versus 
|
The most important geometrical parameters (i.e., bond lengths and bond angles) involved in hydrogen bonding moieties for 1-enol, 1-spt, and 1-dpt forms are listed in Table 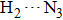
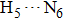


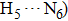

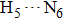


 | Fig. 4. Our theoretical IR spectrum of O1–H2 and O4–H5 stretching vibrational modes for 1-enol structure in S0 and S1 states based on DFT and TDDFT methods, respectively. |
| Table 1.
Simulated bond distances (in unit Å) and bond angles (°) involved in hydrogen bonding moieties for 1-enol, 1-spt, and 1-dpt forms in both S0 and S1 states based on the DFT/TDDFT method in DMSO solvent, respectively. . |
Furthermore, it cannot be denied that hydrogen bonding energy should be a more effective and visual physical quantity to check the variation between S0 and S1 states. Therefore, we also calculate the hydrogen bonding energy in S0 and S1 states for 1-enol system. Hydrogen bonding energy is calculated by subtracting the energy of 1-open configuration from the energy of 1-enol structure. In fact, it is imperative to emphasize at this stage that the estimation of the intramolecular hydrogen bonding energy according to the energy difference between the 1-enol and 1-open configurations produced by the rotation of the twist angle supports the assumption of no other geometry effects as a result of the rotation for hydroxyl O–H. Even though the assumption is perhaps inadequate for simulating the intramolecular hydrogen bonding energy, previous work has proved the feasibility of this method for comparing bonding energy approximately.[62–64] The simulated hydrogen bonding energy of 1-enol in the S0 state is about 8.35 kcal/mol, while that of 1-enol in the S1 state is 12.17 kcal/mol. Thus, we can confirm that the hydrogen bonding does strengthen in the first excited state, which provides the possibility for the ESIPT process.
To examine the effects originating from photo-excitation process, the vertical excitation process from the S0-state optimized 1-enol configuration is also calculated by using TDDFT with the six low-lying absorbing transitions in DMSO solvent. For convenience, the simulated electronic transition energy values, relative oscillator strengths and compositions of first three transitions are listed in Table 


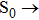

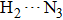

 | Fig. 5. Corresponding frontier molecular orbitals (HOMO and LUMO) for 1-enol system based on TDDFT/B3LYP theoretical level. |
| Table 2.
Theoretical electronic excitation energy (λ nm), corresponding oscillator strengths (f) and corresponding compositions of 1-enol configuration in DMSO solvent for three different transtions. . |
Even though the tendency of ESIPT can be revealed via the above-mentioned analyses, the detailed mechanism is lacking. In fact, the double ESIPT process mentioned is questionable since the energy of the sTable
 | Fig. 6. constructed potential energy profile for S0 and S1 states for 1-enol system, with O1–H2 and O4–H5 bond distances fixed, obtained by DFT and TDDFT methods, respectively. |
4. Conclusions
In this work, we theoretically explore the excited state intramolecular double hydrogen bonding interactions and ESIPT behaviors for the novel 1-enol system. Comparing the geometrical parameters and corresponding IR spectra of hydrogen bonding moieties of 1-enol in both S0 and S1 states, we verify the strengthening of dual intramolecular hydrogen bonds for 1-enol in the first excited state. After investigating the charge distribution and charge transfer resulting from photo-excitation process, we present that the increased electronic densities around proton acceptors play an important role in attracting hydrogen proton. That is to say, the driving force for ESIPT process can be produced by charge redistribution. Further, we construct the PESs of both S0 and S1 states each with an ESIPT pathway. Based on the PESs, we confirm the excited state intramolecular single proton transfer mechanism for 1-enol system. Our work could let researchers have an in-depth understanding of the excited state behaviors involved in multiple hydrogen bonding interactions, and also figure out the detailed ESIPT mechanism for the novel 1-enol system. We expect that this work could play a role in paving the way for revealing and developing novel applications based on 1-enol molecule in future.
Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] | |
| [33] | |
| [34] | |
| [35] | |
| [36] | |
| [37] | |
| [38] | |
| [39] | |
| [40] | |
| [41] | |
| [42] | |
| [43] | |
| [44] | |
| [45] | |
| [46] | |
| [47] | |
| [48] | |
| [49] | |
| [50] | |
| [51] | |
| [52] | |
| [53] | |
| [54] | |
| [55] | |
| [56] | |
| [57] | |
| [58] | |
| [59] | |
| [60] | |
| [61] | |
| [62] | |
| [63] | |
| [64] |