




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Forward-headed structure change of acetic acid–water binary system by stimulated Raman scattering
Cite this Article
Liu Zhe, Yang Bo, Zhao Hong-Liang, Li Zhan-Long, Men Zhi-Wei, Wang Xiao-Feng, Wang Ning, Cao Xian-Wen, Wang Sheng-Han, Sun Cheng-Lin. Forward-headed structure change of acetic acid–water binary system by stimulated Raman scattering. Chinese Physics B, 2019, 28(9): 094206 
Permissions
Forward-headed structure change of acetic acid–water binary system by stimulated Raman scattering
† Corresponding author. E-mail:
Abstract
Abstract
The acetic acid–water binary system is a classical hydroxy–carboxy mixed system, while new and interesting phenomena appear under stimulated Raman scattering (SRS). Compared with the weaker signal of the acetic acid–water binary system obtained in spontaneous Raman scattering, SRS provides a finer band and a relatively distinct structural transition point. The structural transformation points are respectively at 30% and 80% by volume ratio under the condition of spontaneous Raman spectroscopy, while they are respectively at 15% and 25% under the condition of SRS. This phenomenon is attributed to the generation of laser induced plasma and shockwave induced dynamic high pressure environment during SRS.
1. Introduction
Acetic acid, the main component of vinegar as well as one of the most important organic acids, is widely used in pesticides, medicine, organic chemical industry, food additives, and other fields.[1–5] Investigation about the structural feature of vinegar–water mixed solution has always been the focal point in the purification of vinegar.[6–8] Based on previous researches, the main factor affecting the separation efficiency of vinegar out of water is the various hydrogen bond (H bond) structures in the acetic acid–water solution.[9,10] Generally speaking, H bonding is a kind of special intermolecular interaction, existing in systems with a large number of H molecules (generally liquid phase), and plays an important role in many physical properties of liquid substances.[11–13] In the acetic acid–water solution, the acetic acid molecule is both the donor and the acceptor of H bond simultaneously. Two or more H bonds combination modes of acetic acid molecules and water molecules result in different molecular association structures.[14,15] The molecular association of acetic acid affects various physical and chemical properties of the acetic acid–water solution directly. Accordingly, this topic has always been a research hot spot worldwide. Jakobsen et al. considered that the main structure of liquid acetic acid is a cyclic dimer composed of two O–H 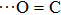


Spontaneous Raman spectroscopy shows relatively broad and weak peaks, bringing about difficulties to the correlated researches. Stimulated Raman scattering (SRS) spectroscopy, as a supplement of spontaneous Raman spectroscopy, has gradually entered the material analysis field.[19] SRS is usually excited by high-energy pulsed laser which can be used to study the molecular structure under the extreme conditions of instantaneous high temperature and high pressure. The Raman peaks in SRS are sharper, so the cluster structure of liquid molecules can be studied more accurately. After the intense pulsed laser beam is focused in the material, the energy density at the focal point increases instantaneously. When the energy density is larger than the threshold of the material, plasma generates.[20] Finally, the internal environment of the material changes dramatically. With the propagation of the laser induced plasma, a large number of excess electrons generate as well. The electrostatic effect of MW order of magnitude can be formed in the local area of the material.[21] Therefore, SRS spectroscopy is often applied in the study of H bonding structures in aqueous solutions due to the enhanced Raman signals. Recent years, the SRS method goes through a rapid development in the field of H bonding network in liquid water thanks to continuous efforts of researchers. Yuan et al. excited two abnormal radiations at 460 nm and 480 nm respectively by 532 nm nanosecond laser, which were caused by the anti-Stokes phenomenon of SRS at the water–air or water–plasma interface.[22] Kumar et al. discovered the energy-dependent evolution of Stokes and anti-Stokes lines of ice VII in liquid water using a strong pulsed laser with a wavelength of 532 nm and a width of 30 ps.[23] Hafizi et al. demonstrated that the Stokes wave could re-focus the pump wave after the power falls below the critical power through an analytical model and propagation stimulation.[24] Our group also investigated the strong and weak H bonds in liquid water and heavy water with the pre-resonance enhancement SRS under dynamic high pressure.[25,26] SRS method plays an important role in the study of H bond structure in liquid water, however, little has been used in the study of acetic acid and acetic acid–water solution.
Herein, both the spontaneous Raman spectra and the SRS spectra of acetic acid and its aqueous solution under different blending ratios were discussed in depth. Different results were found between the two kinds of Raman spectra. A two-peaks distribution of acetic acid and water occurred at a volume ratio of 15% in the SRS spectra, which was different from the corresponding spontaneous Raman results. We analyzed the spectral characteristics of different volume ratios of acetic acid–water solution from the view of the corresponding H bond and acetic acid–water cluster structure. This research will be helpful to improve the separation efficiency of vinegar–water on the basis of molecular vibration.
2. Experiments
Ultra pure liquid water and analytically pure acetic acid were employed to prepare the acetic acid–water binary system, which both were purchased from Sigma-Aldrich Company. The spontaneous Raman spectra of the acetic acid–water binary system were measured and recorded by an InVia Raman confocal microscopic spontaneous Raman spectrometer manufactured by Renishaw. The 514.5 nm laser source with an output power of 2.2 mW was focused on the sample with a 20 × magnification objective lens. The integration time is 60 s and the resolution is 1 cm−1.
In the SRS experiment, a frequency-tripled Nd:YAG laser emited a laser pulse with 8 ns width at a wavelength of 355 nm in base mode. The pulsed laser beam was focused by a focal lens (f = 20 cm) into the acetic acid–water solution in a quartz cell of 100 mm × 10 mm × 45 mm. The forward SRS signal was received by a Maya 2000 spectrometer. All the experiments were carried out continuously under normal temperature and pressure.
3. Results and discussion
First, the spontaneous Raman spectra of the acetic acid–water solution with different volume ratios were measured. The volume ratio of acetic acid varied from 10% to 100% in steps of 10%. As shown in Fig.
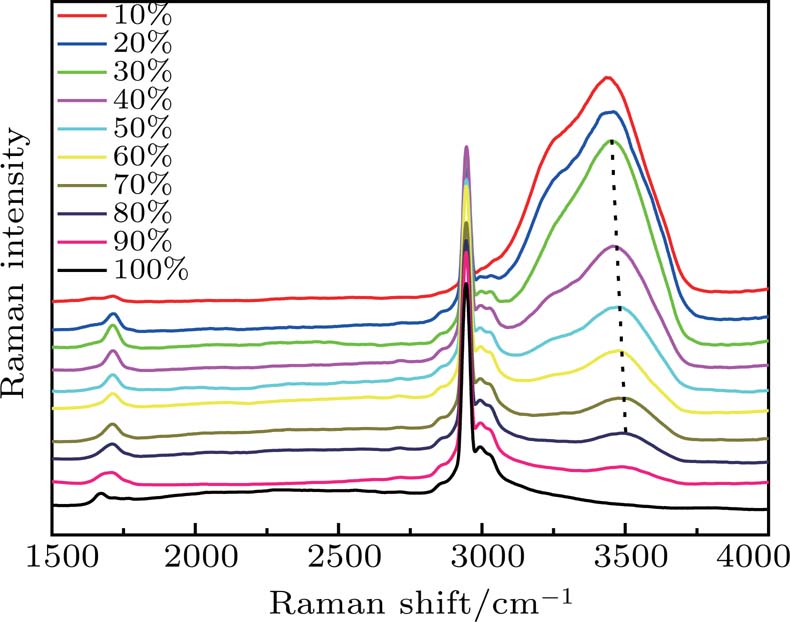 | Fig. 1. Spontaneous Raman spectra of acetic acid–water solution with different volume ratios. |
 | Fig. 2. (a) Raman spectra of OH vibration of acetic acid–water solution from 3000cm−1 to 3800 cm−1 in different blending ratios and (b) the corresponding variation trend of Raman shift of bands at 3242 cm−1 and 3443 cm−1. |
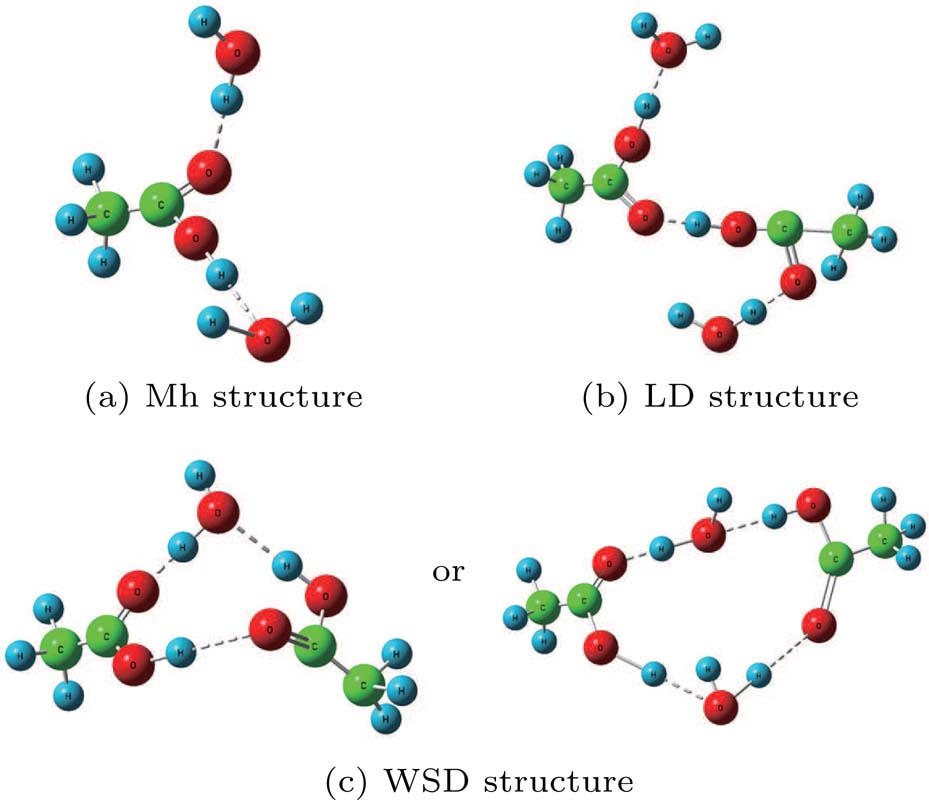 | Fig. 3. Three kinds of molecular structures in acetic acid–water solution: (a) Mh structure, (b) LD structure, (c) WSD structure. |
As shown in Fig.
 | Fig. 4. (a) Raman spectra of C=O stretching vibration of acetic acid-water solution from 1600 cm−1 to 1800 cm−1 in different blending ratios and (b) the corresponding variation trend of Raman shift of band at 1714 cm−1. |
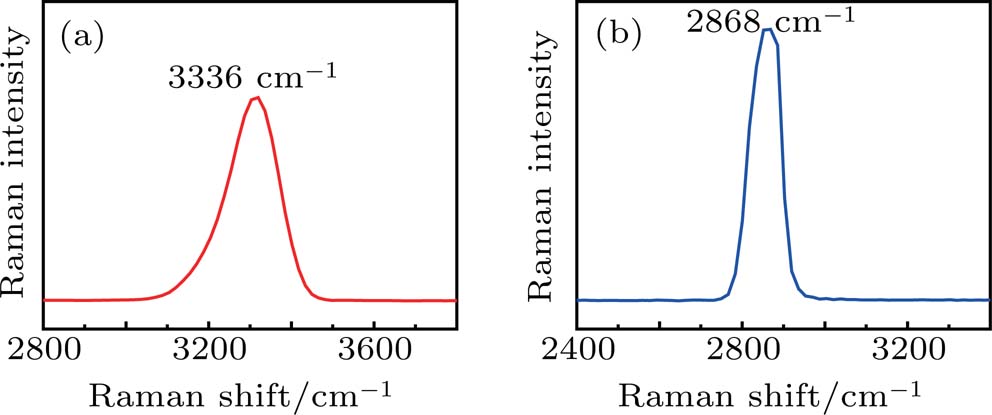
Apart from spontaneous Raman measurements, SRS features of the acetic acid–water solution were measured in different blending ratios. Compared with the abundant vibration distribution of spontaneous Raman spectra, only two vibration peaks were detected by SRS, namely, the CH3 stretching vibration peak at 2868 cm−1 of acetic acid and the OH vibration peak at 3336 cm−1 of water. This phenomenon is caused by the different excitation mechanisms of SRS and spontaneous Raman scattering. Under the condition of steady state limit, the optical path length in the medium is defined as z, then the intensity of the output Stokes signal can be expressed as
 | Fig. 5. Spectrum of acetic acid-water solution when the volume ratio is (a) less than 15% and (b) more than 25%. |
The SRS spectra are shown in Fig.
 | Fig. 6. SRS spectra of acetic acid–water solutions with different volume ratios. |
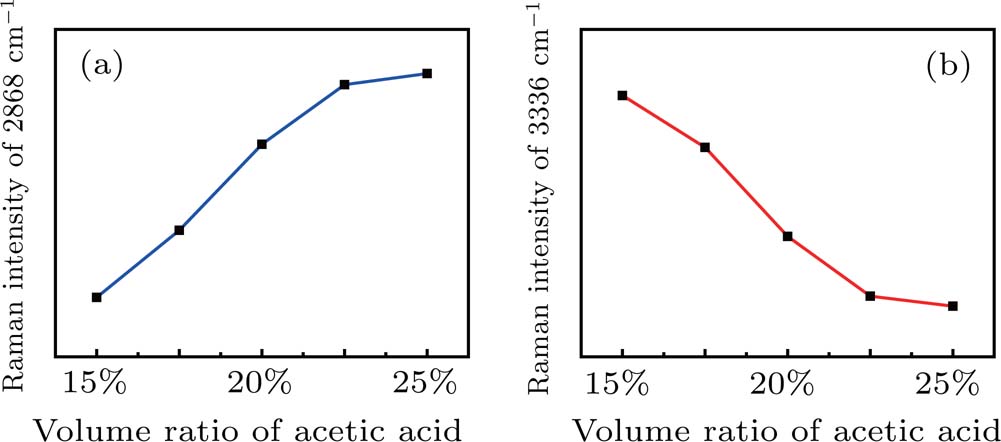
 | Fig. 7. Intensity variation of Raman peaks with volume ratio: (a) 2868 cm−1 and (b) 3336 cm−1. |
Except for the change of the Raman peak intensity, the Raman shifts corresponding to the OH stretching vibration peaks at 3316 cm−1, 3330 cm−1, 3336 cm−1, and 3336 cm−1 show a trend of moving towards high wavenumber as the volume ratio increases from 15% to 22.5%. The main reason for this phenomenon is that the proportion of LD structure increases gradually. Compared with the Mh structure, the H bond strength of water in the LD structure is relatively large, which will cause the OH bond to be stretched. Thus, the corresponding peak moves towards the high wavenumber. In conclusion, the peak position moves to high wavenumber as the volume ratio increases from 15% to 22.5% in the SRS experiment.
The structural change points of SRS are 15% and 25%, while those of the spontaneous Raman scattering are 30% and 80%. Distinct differences can be seen from the results. The main reason is that the laser induced plasma and the followed shockwave induced dynamic high pressure will be generated in the SRS process. Under the shockwave induced dynamic high pressure, the cluster structure of water changes in advance. After the intense laser pulse is focused in the liquid water, the laser energy at the focal point is larger than the breakdown threshold of water. A laser induced breakdown (LIB) phenomenon at the focal point of the excitation beam is produced. A plasma cloud which contains a large number of nanobubbles near the ionization region forms by absorbing the laser energy.[40] Since the pressure inside the bubbles is far higher than outside, the plasma nanobubbles burst instantaneously, resulting in a strong shock wave inducing GPa-level dynamic high pressure region. An electric field generates while excess electrons continue to propagate in the dynamic high pressure region. This extreme environment has an impact on the H bonding structure and OH vibration, leading to the structural change points of acetic acid–water solutions moving forward.
4. Conclusion
In summary, the variation of H bond network of the acetic acid–water binary system with different volume ratios is investigated detailedly by spontaneous Raman scattering and SRS methods. Both of spontaneous Raman scattering and SRS spectra demonstrate the existence of two structural transformation points in the acetic acid–water binary system with different volume ratios. However, the SRS results exhibit different features from the spontaneous Raman ones due to the different excitation mechanisms. Great emphases have been put on the laser-induced plasma produced by SRS and the accompanying shockwave induced dynamic high pressure environment. The dynamic effect of this phenomenon on the acetic acid–water binary system is also analyzed. This paper contributes to extend the application of SRS spectroscopy in the area of liquid materials and guide significance for the purification of acetic acid.
Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] | |
| [33] | |
| [34] | |
| [35] | |
| [36] | |
| [37] | |
| [38] | |
| [39] | |
| [40] |