




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Structural model of substitutional sulfur in diamond
Cite this Article
Yu Hongyu, Gao Nan, Li Hongdong, Huang Xuri, Duan Defang, Bao Kuo, Zhu Mingfeng, Liu Bingbing, Cui Tian. Structural model of substitutional sulfur in diamond. Chinese Physics B, 2019, 28(8): 088102 
Permissions
Structural model of substitutional sulfur in diamond
† Corresponding author. E-mail:
Abstract
Based on ab initio calculations, it is found that the donor center of substitutional sulfur (S) in diamond with 




Keyword:substitutional sulfur in diamond;structural model;molecular dynamic simulation;supercell size
1. Introduction
Diamond having unique physicochemical properties is considered as one of the most promising materials for next generation semiconductors.[1] It is well-known that p-type diamond have been successfully obtained by boron (B) doping,[2,3] however, the fabrication of n-type diamond with a shallow donor level is extremely difficult. In order to achieve the shallow doping for n-type diamond, kinds of donors have been attempted, such as lithium,[4,5] nitrogen,[6–9] phosphorus,[10–13] oxygen,[14] and sulfur (S),[15–17] etc.
Doping with S was reported to be an effective method to obtaining the n-type diamond,[15,18] then a number of computational approaches have been employed to estimate the donor model and donor level value.[19–31] One possibility of the origin of the donor levels may be substitutional S (Ssub),[1] and the most stable configuration of Ssub in diamond is generally considered to S atom and four carbon (C) neighbors with 


Since the molecular dynamic (MD) simulations can exhibit the changing of the structure parameter of Ssub in diamond at room temperature as the function of time, it has been successfully used to predict the new structure of ammonium azide under high pressure,[32] consider the tanglesome rotation of ammonium cation in ammonium nitrate[33] and understand the kinetic and thermodynamic properties of the closing and opening of a base pair.[34] In this paper, both the ab initio MD simulations and structural optimizations are considered for the structural model of Ssub in diamond with the larger supercells. We find that the 
2. Computation details
All calculations performed in this paper are based upon ab initio density functional theory simulations, as implemented in Vienna Ab-initio Simulation Package (VASP) code.[35] The projector-augmented wave (PAW)[36,37] method is adopted and the cutoff energy of 520 eV is set. The Perdew–Burke–Ernzerhof (PBE) function based on the generalized gradient approximation (GGA)[37] is employed to describe the exchange and correlation effects of electrons. The MD simulations are implemented both in the isobaric isothermal (NPT) ensemble at 300 K, and atmospheric pressure and canonical (NVT) ensemble at 300 K. The optimized lattice parameters of pristine diamonds with 64-, 216-, and 512-atom supercells are 7.146 (3.573 × 2), 10.7189 (3.573 × 3), and 14.292 (3.573 × 4) Å, which are agree well with the experimental single cell lattice value of 3.567 Å,[38] and then one C atom is replaced with a S atom for MD simulations. In MD simulations, the Brillouin zone is sampled using the Monkhorst–Pack scheme, with mesh of 3 × 3 × 3, 2 × 2 × 2, and 1 × 1 × 1 for 64-, 216-, and 512-atom supercells, respectively. The time steps of 1.0 fs and total simulation times of at least 10 ps are used, and all the MD simulations achieve thermalisation after 3 ps by examining the system pressure and temperature. After MD simulations, we optimize the averaged structures and calculate the total energies of the three supercells with k mesh of 7 × 7 × 7, 5 × 5 × 5, and 3 × 3 × 3, respectively.
3. Results and discussions
First, the MD simulations both in the NVT and NPT ensembles at 300 K are performed to investigate the Ssub structure. The main difference between the NVT and NPT ensembles is that the volume, side length and side angle of supercell for the former are restricted constant, while those in the latter are allowed to optimize. Figure 


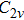


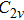
 | Fig. 1. The distance between S and four adjacent C atoms (d_SCi, i = 1, 2, 3, 4) as a function of the MD simulation time in (a) NVT and (b) NPT ensembles, and the six bond angles in donor center (
|
Figure 









 | Fig. 2. The distance between S and four adjacent C atoms (d_SCi, i = 1, 2, 3, 4) as a function of the MD simulation time in (a) NVT and (b) NPT ensembles, and the six bond angles in donor center (
|
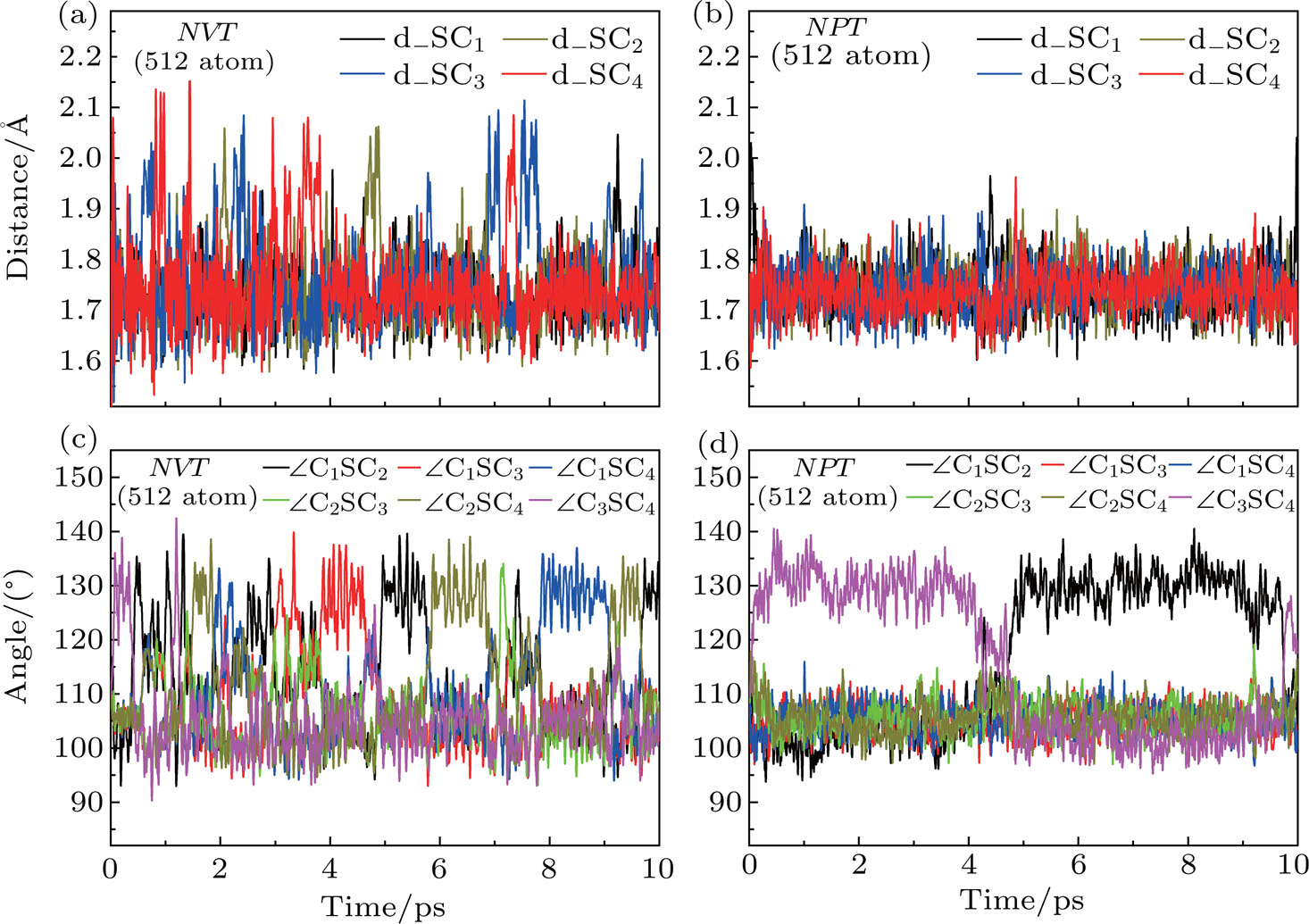 | Fig. 3. The distance between S and four adjacent C atoms (d_SCi, i = 1, 2, 3, 4) as a function of the MD simulation time in (a) NVT and (b) NPT ensembles, and the six bond angles in donor center (
|
To explain it, the three lattice angles of Ssub in diamond with 64-, 216-, and 512-atom supercells and pristine diamond with 512-atom supercell during NPT MD simulations are given in Fig.
 | Fig. 4. The lattice angle of Ssub in diamond with (a) 64-, (b) 216-, and (c) 512-atom supercells and (d) pristine diamond with 512-atom supercell as a function of NPT MD simulation time. |
Then, the structures under a series of density functional theory (DFT) structural optimizations are obtained to further confirm the stable Ssub structure. First, the ionic positions, the lattice shape and parameters of 













| Table 1.
The lattice parameters of fully relaxed supercells with |
| Table 2.
The parameters of relaxed cubic supercells with |
The band structures of 512-atom with 




 | Fig. 5. The band structures of Ssub in diamond with 512-atom supercell with 
|
 | Fig. 6. Partial structures of [(a), (b), (c)] ELF and [(d), (e), (f)] charge density of Ssub in diamond in 512-atom supercell with 
|
4. Conclusions
In conclusion, the stable donor center structure of Ssub in diamond is studied by a series of MD simulations and structural optimizations using the 64-, 216-, and 512-atom supercells. The 


Acknowledgment
The calculations were performed in the High Performance Computing Center (HPCC) of Jilin University.
Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] | |
| [33] | |
| [34] | |
| [35] | |
| [36] | |
| [37] | |
| [38] | |
| [39] | |
| [40] |