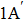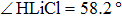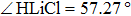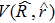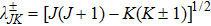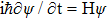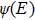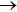Zhai Hongsheng, Liang Guanglei, Ding Junxia, Liu Yufang. Isotope effect and Coriolis coupling effect for the Li + H(D)Cl → LiCl + H(D) reaction. Chinese Physics B, 2019, 28(5): 053401
Permissions
Isotope effect and Coriolis coupling effect for the Li + H(D)Cl → LiCl + H(D) reaction
College of Physics and Materials Science, Henan Normal University, Xinxiang 453007, China
Spectral Measurement and Application of Infrared Material Key Laboratory of Henan Province, Academy of Physics and Materials Science, Henan Normal University, Xinxiang 453007, China
State Key Laboratory of Molecular Reaction Dynamics, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
A time-dependent quantum wave packet method is used to investigate the dynamics of the Li+ H(D)Cl reaction based on a new potential energy surface (J. Chem. Phys.146 164305 (2017)). The reaction probabilities of the Coriolis coupled (CC) and centrifugal sudden (CS) calculations, the integral cross sections, the reaction rate constants are obtained. The rate constants of the Li+ HCl reaction are within the error bounds at low temperature. A comparison of the CC and CS results reveals that the Coriolis coupling plays an important role in the Li+ H(D)Cl reaction. The CC cross sections are larger than the CS results within the entire energy range, demonstrating that the Coriolis coupling effect can more effectively promote the Li+ DCl reaction than the Li+ HCl reaction. It is found that the isotope effect has a great influence on the title reaction.
As a prototype for the three-atom reactions between alkali metals and hydrogen halides, the reaction Li+ HCl has been the subject of various theoretical and experimental investigations.[1–14] On aspects of experiments, Helbing and Rothe[1] measured the velocity dependence of the total cross section integral cross sections for the scattering of lithium by a series of molecules (include the HCl and DCl molecules). In Becker's[2] group, the molecular beam method was adopted to study the reaction of Li+ HCl, the results showed that the majority of the energy in this reaction goes to the translational energy of the product, and only a little energy is transferred to the vibration energy of the product. This reaction's barrier is lower than 2.00 kcal/mol, and there is a van der Waals well at the entrance. After that, Plane and Saltman[3] measured the reaction rate constants by the technique of time-resolved laser induced fluorescence spectroscopy of Li between 700 K and 1000 K. The height of this reaction's barrier is about 1.96 ± 1.4 kcal/mol.
On the theoretical side, in 1979, the first potential energy surface (PES) was constructed by Shapiro and Zeiri[4] using a semi-empirical diatomic-in-molecules (DIM) approach. The transition state derived from the semi-empirical calculations is located at the entrance channel and has collinear geometry. Its transition state energy barrier is 11.36 kcal/mol. Garcia et al.[5] presented a dynamical investigation of the Li+ HCl reaction carried out on the DIM potential energy of Zeiri and Shapiro.[6] The threshold energy estimated from these calculations conflicts with the large value of the reactive cross section measured using a crossed molecular beams apparatus. Garcia and Lagana[7] reported the configuration interaction (CI) computation of the potential energy at several geometries of the LiHCl system and the computations exhibit a bent structure for the transition state. Subsequently, Palmieri et al. constructed a LiClH PES fitted by the bond order (BO)[8] polynomial and the new BO PES by modifying the ab initio points.[9] Based on the new BO PES,[10] the rate constants and cross sections were calculated, and the rate constants obtained from the new BO PES were compared with the experimental results of time-resolved laser induced fluorescence spectroscopy. The dynamics calculations indicate that the higher quality ab initio PES is needed.[11–15] In recent years, Chen et al.[16] and Lin et al.[17] obtained high-precision global PES crucial for the LiHCl system ground state, respectively. And Lin's group reported the integral cross sections, rate constants, and their dependence on the initial rotational states based on their new PES. The rate constants are in agreement with the experimental results.
Coriolis coupling effects can influence dynamical properties of polyatomic molecular reactions, and have been a subject of experimental and theoretical investigations.[18–26] Coriolis coupled (CC) item is implemented by the time-dependent wave packet method with a split-operator scheme.[24,29] In order to analyze the Coriolis coupling effect, the centrifugal sudden (CS) approximation that ignores Coriolis coupling is usually employed in the dynamics calculations.[22,23] For this work, both CC and CS calculations for the title reaction are implemented.[17] The rest of this paper is arranged as follows. Section 2 presents the new ab initio PES and the method of dynamics calculation. Section 3 analyses the effect of the isotope and Coriolis coupling on the title reaction, and section 4 provides the conclusions.
2. The characteristics of the PES and calculation method
The lowest adiabatic potential energy surface for the Li+H(D)Cl reaction[17] is constructed with the three-dimensional cubic spine interpolation method by fitting the multi-reference configuration interaction calculations on a large basis set. As described in the literature,[17] the title reaction is exothermic. There are two very shallow van der Waals wells (−0.645 kcal/mol and 0.68 kcal/mol) at the reactant channel, one low energy barrier (2.99 kcal/mol) at the location (RHCl=2.68 Bohr, RLiCl=4.29 Bohr, , and one well (−8.35 kcal/mol) for the complex structure near the product channel (RHCl=4.02 Bohr, RLiCl=3.956 Bohr, ). The reaction is likely dominated by a direct mechanism.
The quantum dynamics calculation is carried out with the time-dependent wave packet method developed by Zhang et al.[28,29] The Hamiltonian of the LiHCl reactive system can be expressed as
where R is the distance between the Li atom and the center-of-mass of HCl, r is the HCl bond length, μR is the reduced mass of Li with respect to HCl, μr is the reduced mass of HCl, J is the total angular momentum, j is the rotational angular momentum of HCl, is the interaction potential, and h(r) is the diatomic reference Hamiltonian
with V(r) being the diatomic reference potential. The initial wave packet is structured with the body-fixed (BF)[21] translational vibration rotational basis.
The element of the centrifugal term or the CC matrix is expressed as
with . In the CS approximation,[22] the off-diagonal terms are neglected. Then the above equation can be simplified as
The initial Gaussian wave packet can be written as the product of the motion wave packet and the initial vibration Eigen-function. The time-dependent Schrödinger equation () is used in the reactant Jacobi coordinates by the split-operator scheme as follows:
where the reference Hamiltonian H0 is defined as
and the effective potential operator U is defined as
The reaction probability, the integral reaction cross sections, and the temperature dependent rate constant are expressed as
Here, is the final wave function, KB is the Boltzmann constant, E is the collision energy, and σ (E) is the fitted integral cross section.
3. Results and discussion
Table 1 shows the dynamical parameters used in the quantum calculations for this work. In order to obtain better convergence results, firstly, the number of base functions is selected. 383 cell points are divided by 2.2–20.0 a0 at the R coordinate, 199 points split the 1.2–16.0 a0 at the r coordinate, and jmax =110. Then the related parameters in the initial wave packet are determined. The initial wave packet with a width of 0.32a0, an average translational energy of 0.14 eV, and propagation time 45000 is long enough. Finally, the absorption potential (3.0/0.03) is introduced to prevent the wave packet from bouncing at the boundary.
Table 1.
Table 1.
Table 1.
Parameters for the calculations in this work (all quantities are given in units of a.u., unless otherwise indicated).
.
Range of collision energy/eV
0.05–0.7
Width of the initial wave packet
0.32
Average translational energy/eV
0.14
Coordinate range of R
2.2–20.0
Lattice of translational basis functions
383
Lattice of vibration basis functions
199
Max for the rotational basis functions jmax
110
Coordinate range of r
1.2-16.0
Run time
45000
Absorption potential
3.0/0.03
Table 1.
Parameters for the calculations in this work (all quantities are given in units of a.u., unless otherwise indicated).
.
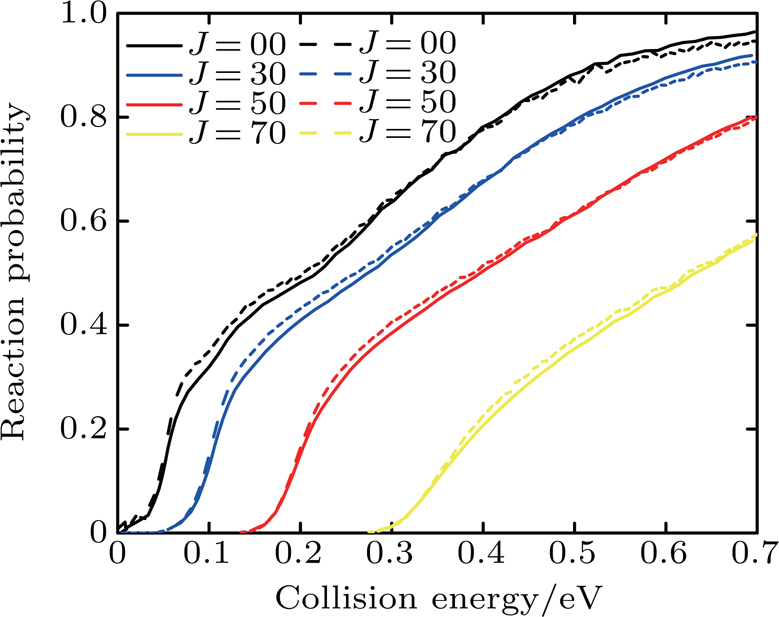
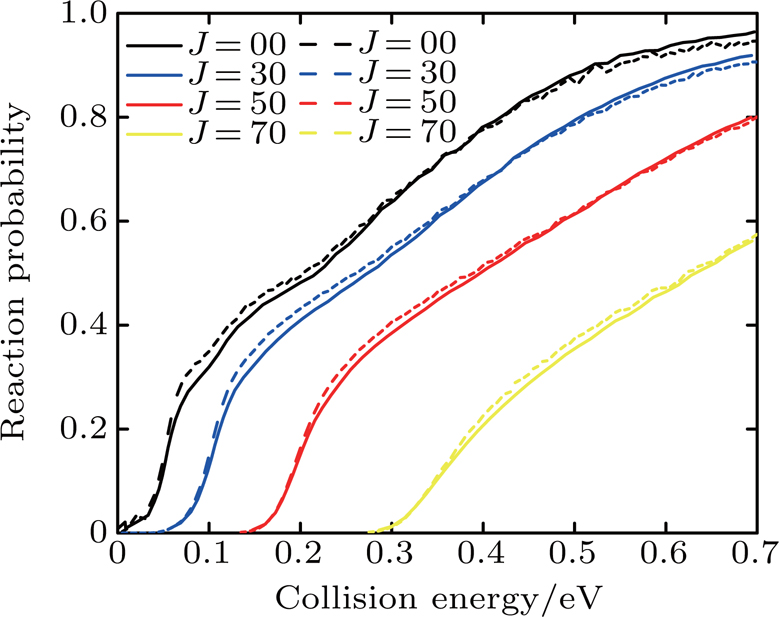
Figure 1 compares the dependence of the reaction probabilities with different J (0, 30, 50, 70) on the collision energy of the Li+ HCl (v = 0, j = 0) reaction between the calculations of Lin et al.[17] and this work. Although our calculation results are slightly higher than those of Lin's group at low collision energy and small J, the two reaction probabilities obviously agree well with each other.
Fig. 1. Dependence of the reaction probabilities with different J (0, 30, 50, 70) on the collision energy of the Li+ HCl (v = 0, j = 0) reaction. The results of Lin et al.[17] (solid line) and this work (dashed line) are shown.
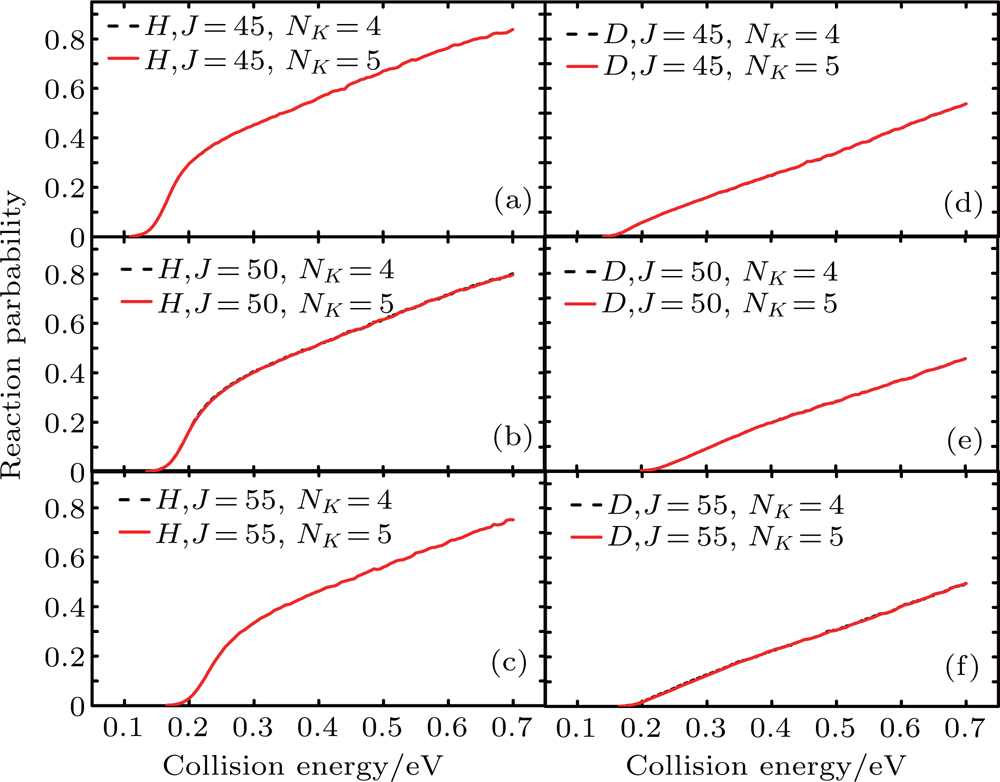
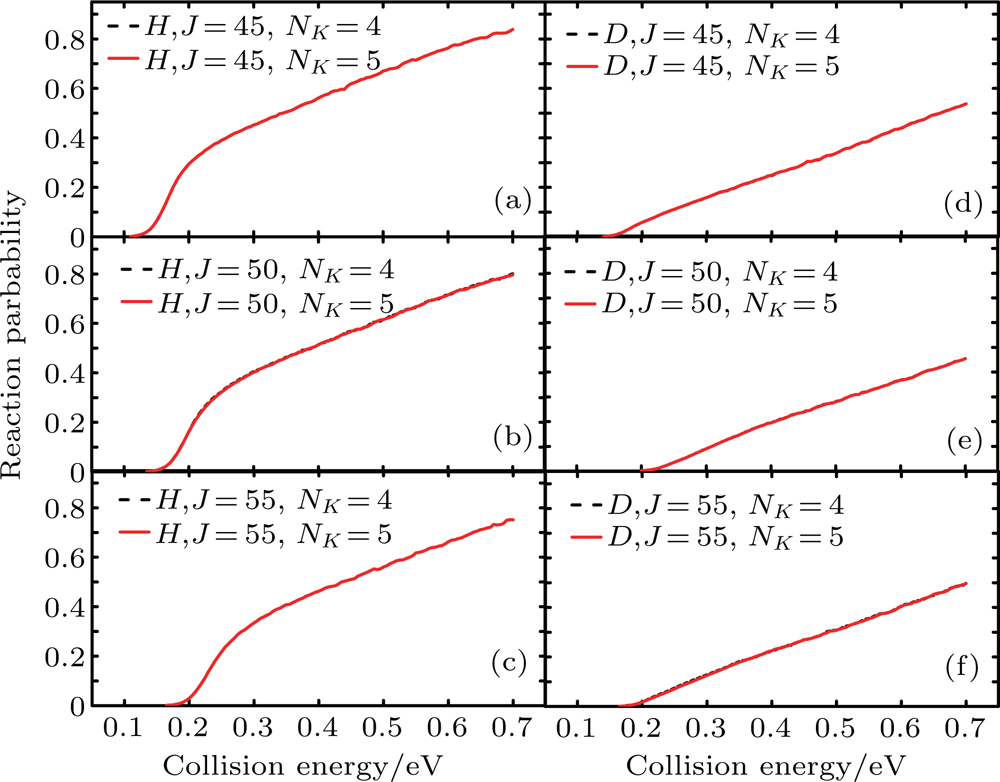
In Fig. 2, the reaction probabilities of the Li+H(D)Cl (v = 0, j = 0) reaction at J = 45, 50, and 55 with NK=4 and 5 are shown, where NK is the number of K states used in CC calculation. Limited by the computational resources and time, convergent NK is required in precise calculations. As shown in Fig. 2, there are little differences in probabilities between NK=4 and NK=5 at higher total angular momentum (J = 45, 50, 55). To save computation time and resources without sacrificing calculation accuracy, NK=4 is used. On this basis, the Coriolis coupling effect of the title reaction is calculated.
Fig. 2.NK-dependent reaction probabilities of the Li+H(D)Cl (v = 0, j = 0) reaction at J = 45, 50, and 55 for the CC calculation.
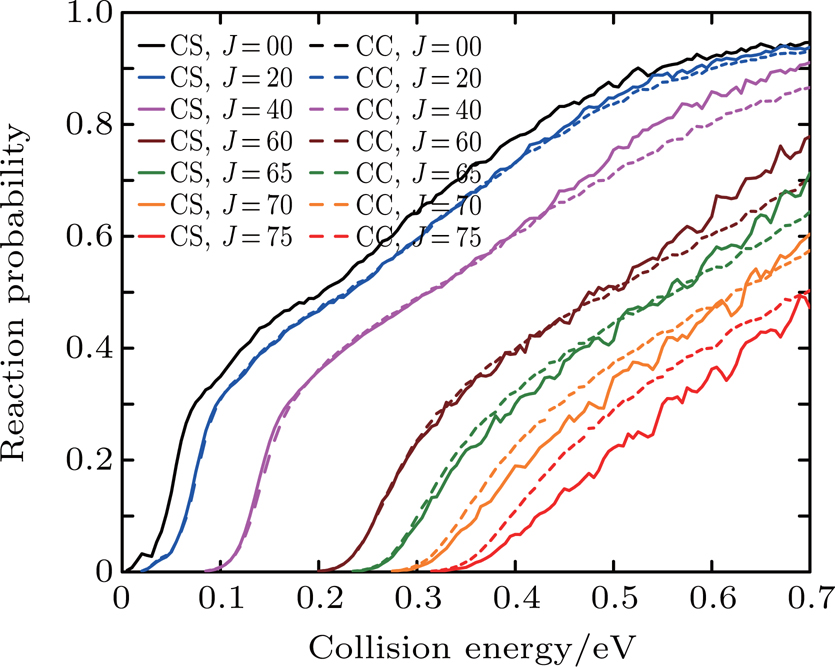
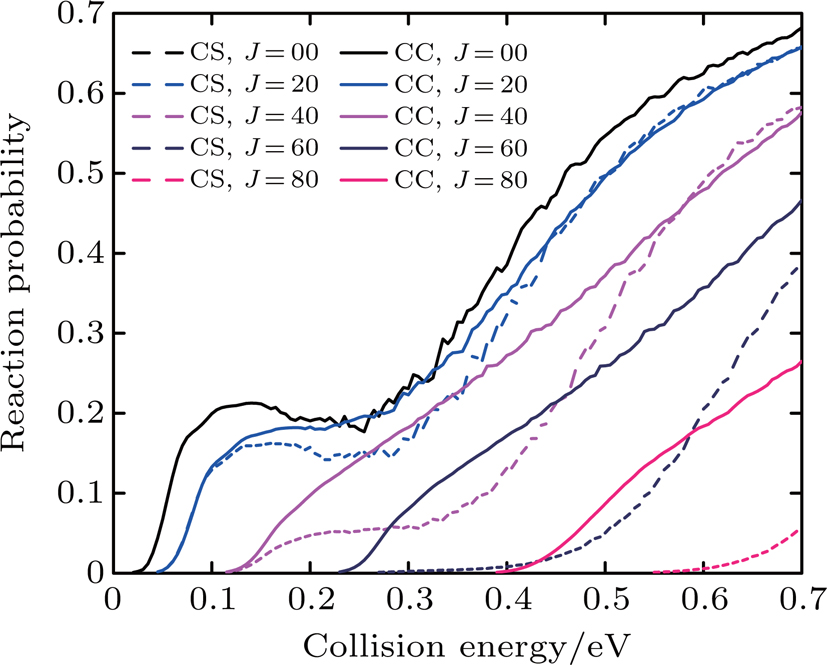
Figure 3 shows the CC and CS probabilities for the initial quantum numbers v = 0, j = 0 at J = 0, 20, 40, 60, 65, 70, and 75 of the Li+ HCl reaction. It can be found that for small J and low collision energy, the differences between the CC and CS probabilities are very little. Only at the high collision energy, the CC results are lower than the CS ones. The remarkable differences are observed with increasing J. At J = 60, the CC result is higher than the CS one at the collision energy from 0.25 eV to 0.35 eV, but lower than the CS one at the high collision energy. As J keeps increasing, the CC calculation results are enhanced, and exceed the CS ones at the whole collision energy region. From the above discussion, we find that the centrifugal potential barrier plays an important role in the Li+ HCl reaction. The differences between the CS and CC results become apparent as the collision energy increases.
Fig. 3. Dependence of the reaction probabilities on the collision energy of the Li+ HCl (v = 0, j = 0) reaction at J = 0, 20, 40, 60, 65,70, and 75.
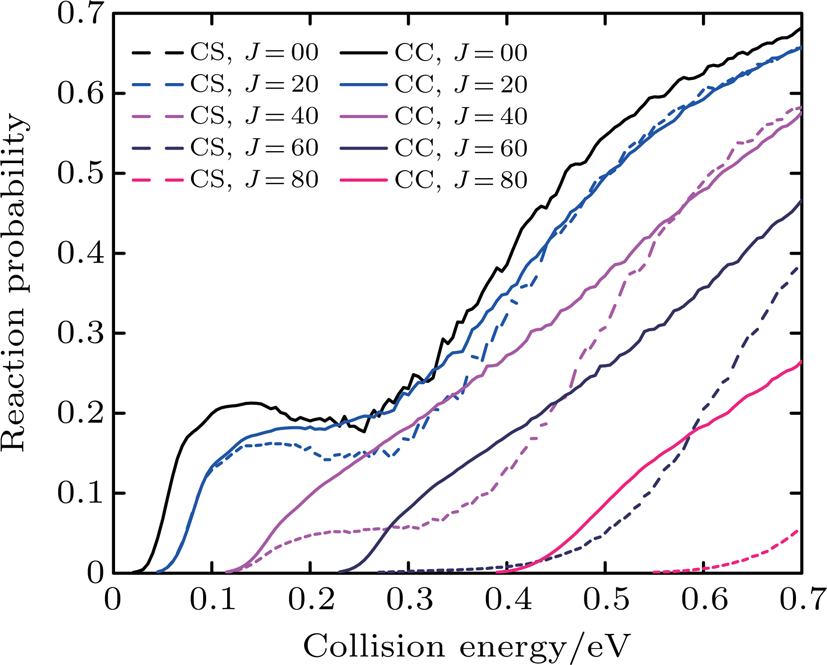
Figure 4 presents the reaction probabilities versus the collision energy of Li+ DCl (v = 0, j = 0) reaction at J = 0, 20, 40, 60, and 80. There is a clear distinction between Li+ HCl and Li+ DCl reactions. In Fig. 4, the Li+ DCl reaction probabilities are significantly depressed. At J = 0, the reaction probability has a decrease at 0.2–0.35 eV and then returns to a significant rise with the increase of the collision energy. Different from the Li+ HCl reaction, the Coriolis coupling effect plays a more import role in the Li+ DCl reaction. With the increase of J, the discrepancy between CC and CS becomes larger than that in the Li+ HCl reaction. And the reaction threshold in the CS calculations is lower than that in CC. The shifting of the reaction thresholds is entirely due to the centrifugal barrier. According to the literature[16,17], the Li+ HCl reaction is dominated by a direct reaction mechanism, and the H atom is too light to be impacted away. However, when the H atom is replaced by a D atom, the molecular rotation is more likely to participate in the collision process. Here, the isotope effect has a great influence on the title reaction. On the one hand, it leads to reduction of the reaction probabilities. On the other hand, it causes the Coriolis coupling effect to be remarkable.
Fig. 4. Dependence of the reaction probabilities on the collision energy of the Li+ DCl (v = 0, j = 0) reaction at J = 0, 20, 40, 60, and 80.
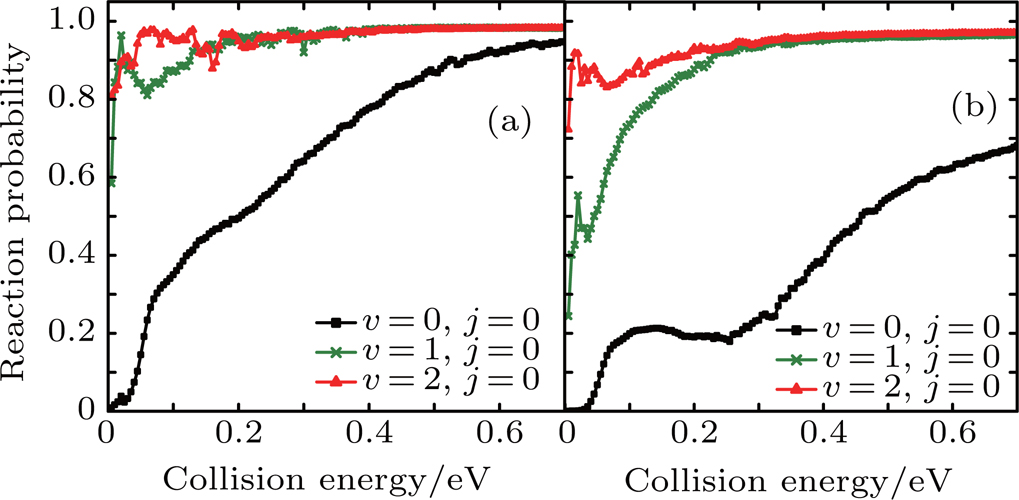
Figure 5 shows the reaction probabilities of Li+ H(D)Cl reaction at j = 0, v = 0–2. The probabilities for v = 1 and 2 are higher than that for v = 0, especially at the low collision energy. Both the probabilities for v = 1 and 2 become close to one. This illustrates that Li+ H(D)Cl LiCl+H(D) is the predominant channel. As the vibrational quantum number v increases, the reaction probability increases. With the increase of the diatom vibration excitation energy, the reactant is easier to cross the barrier, and the reaction productive rate increases accordingly.
Fig. 5. Dependence of the reaction probabilities on the collision energy of the Li+ H(D)Cl (j = 0, v = 0, 1, 2) reaction at J = 0: (a) Li+ HCl, (b) Li+ DCl.
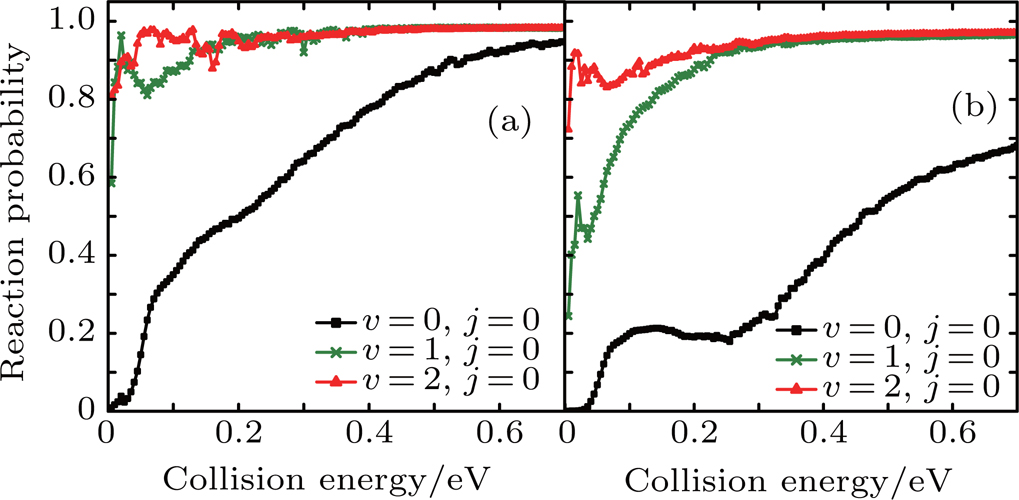
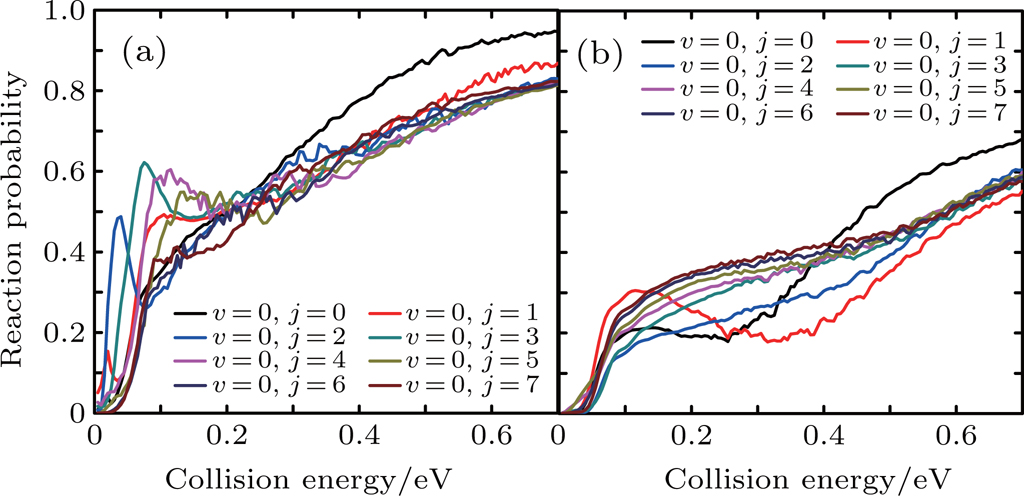
Figure 6 shows the reaction probabilities versus the collision energy of Li+ H(D)Cl reaction at v = 0, j = 0–7. For the Li+ HCl reaction, with higher j, the probabilities show the distinct increase at the low collision energy. When j is greater than 5, this advantage is disappearing. The Li+ DCl reaction has the same dynamical behavior at rotational quantum number j = 0 and 1. Both probabilities decrease in the collision energy region of 0.2–0.4 eV. With the increase of j, the descending disappears and the probabilities rise in the whole collision energy region.
Fig. 6. Dependence of the reaction probabilities on the collision energy of Li+ H(D)Cl reaction at v = 0, j = 0–7: (a) Li+ HCl, (b) Li+ DCl.
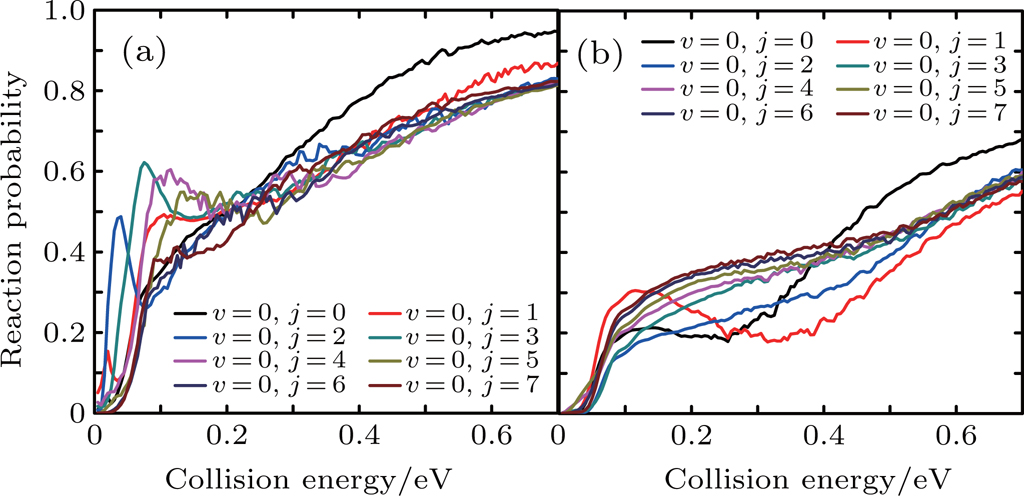
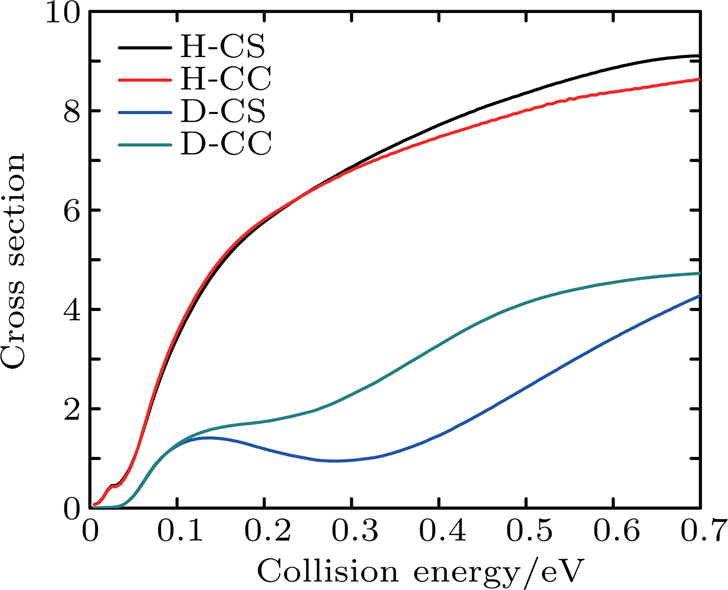
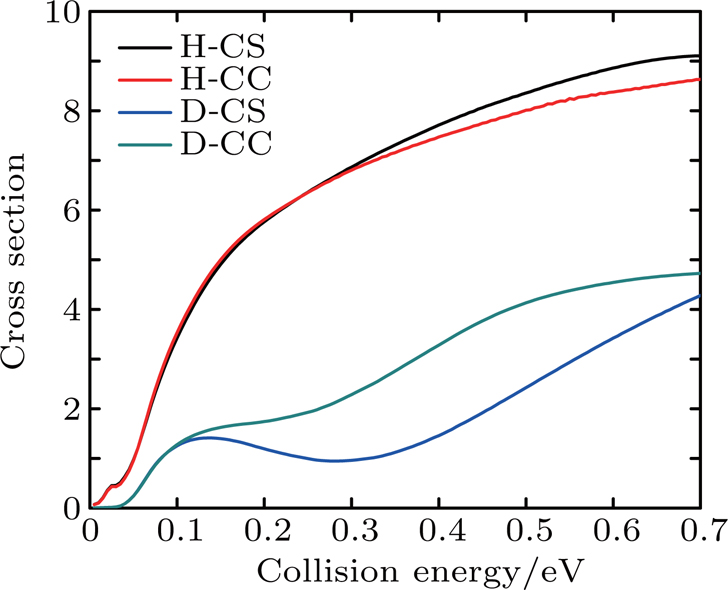
Figure 7 shows the integral reaction cross section calculated by CS and CC for the Li+ H(D)Cl reaction. As can be seen from Fig. 7, the Li+ HCl reaction integral reaction cross sections increase with collision energy. However, for the isotope reaction Li+ DCl, there is a significant depression in the collision energy range of 0.2–0.4 eV, which is consistent with the results of the reaction probability. As is shown above, the CS integral reaction cross section for the Li+ HCl reaction is consistent with the result of CC in the low energy region. As the action on the high J of the Coriolis effect, the CC probability is below the CS result. For the Li+ DCl reaction, due to the heavy quality factor, the Coriolis coupling effect is more obvious. The different of the integral cross section between the CS and CS calculations is outstanding in the collision region of 0.13–0.7 eV. And the CC result is higher than the CS one.
Fig. 7. Integral reaction section for reaction Li+ H(D)Cl.
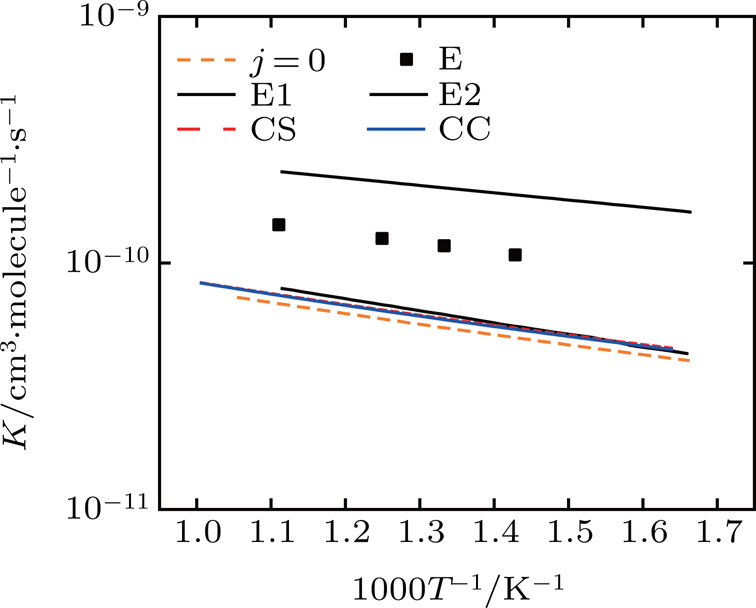
The rate constants of the title reaction are given in Fig. 8. As we can see from the graph, the calculated results of CS and CC from this work are within the error bounds at the low temperature. In the experiment, the HCl molecules are normally in the ground state of vibration and the rotation state of j = 5 is the most distributed.[17] As the literature mentioned,[31,32] the high rotational excitation and vibration excitation can promote the reaction. The probabilities of different j in Fig. 6 also confirm this conclusion.
Fig. 8. Rate constants. The experimental data[30] (black cubes) and its error bounds (black lines), and the literature data of Lin[17] (j = 0, the orange dashed line) are also shown.
4. Conclusion
The CC and CS reaction probabilities as well as the total cross sections are calculated. The differences between the CS and CC results become apparent as the collision energy increases. The lower reaction thresholds of the CC calculations are the main cause of the enhancement of the CC integral cross sections. Therefore, the Coriolis coupling effect can effectively promote the title reaction. The comparison of the calculations between the Li+ HCl and Li+ DCl reactions reveals that the isotope effect has a great influence on the title reaction. On the one hand, it leads to reduction of the probabilities of the Li+ DCl reaction. On the other hand, it causes the Coriolis coupling effect of the Li+ DCl reaction to be remarkable. The probability calculations at v = 0–2, j = 0–7 indicate that the high rotational excitation and vibration excitation can promote the title reaction. The rate constant calculations manifest that the high rotational excitations of the reactant have the positive contribution to the results.
Acknowledgment
The calculation in this work was supported by the High Performance Computing Center of Henan Normal University.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}