

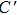

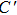
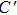
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
First-principles study of structural, mechanical, and electronic properties of W alloying with Zr
Cite this Article
Zhang Ning-Ning, Zhang Yu-Juan, Yang Yu, Zhang Ping, Ge Chang-Chun. First-principles study of structural, mechanical, and electronic properties of W alloying with Zr. Chinese Physics B, 2019, 28(4): 046301 
Permissions
First-principles study of structural, mechanical, and electronic properties of W alloying with Zr
† Corresponding author. E-mail:
Abstract
The structural, mechanical and electronic properties of W1−xZrx (x = 0.0625, 0.125, 0.1875, 0.25, 0.5) are systematically investigated by means of first-principles calculation. The total-energy calculations demonstrate that the W–Zr binary substitutional solid solution remaining bcc structure can be formed at an atom level. In addition, the derived bulk modulus (B), shear modulus (G), Youngʼs modulus (E) for each of W–Zr alloys decrease gradually with the increase of Zr concentration, suggesting that W alloying with higher Zr concentration becomes softer than pure W metal. Based on the mechanical characteristic B/G ratio, Poissonʼs ratio υ and Cauchy pressure 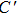
1. Introduction
Tungsten (W) has many applications because it has the highest melting point of all of the elements, it also has a high elastic modulus, high density, high thermal conductivity, and excellent mechanical properties at elevated temperatures, such as in incandescent light bulb filaments, heating elements, and kinetic energy penetrators.[1] The study of W is important for plasma facing material due to its high melting point, high strength at high temperatures and high plasma sputtering erosion resistance.[2–4] However, the poor ductility of tungsten is not conducive to its workability and performance in the applications. Thus, there is an urgent need to improve the mechanical properties of tungsten.
To date, doping impurities and additives in W has been demonstrated to be a promising method to improve physical properties. For example, it is well-accepted that alloying zirconium (Zr) could improve the physical properties of W.[5–8] On the one hand, alloying small amount of element Zr that can bond with oxygen will increase grain boundary strength.[5,6] The incorporation of zirconium by spark plasma sintering in a range of 0.2 wt%–1.0 wt%, with and without additional yttrium oxide (Y2O3) dispersion, have been recently investigated by Liu et al.[5] The results showed that the ductile-brittle transition temperature (DBTT) of W with the addition of 0.2% Zr alone was about 200 °C lower than that of pure W, and ultimate tensile strength was improved by incorporating the Y2O3 particles, which also results in a slight decrease in corresponding ductility. Meanwhile, Xie et al. showed that Zr, decomposed from ZrH2, could capture impurity oxygen in tungsten and form nano-sized monoclinic ZrO2 particles.[6] The tungsten sample with 0.2 wt% ZrH2 addition exhibits not only the highest fracture strength but also the highest toughness. However, the greater than 0.2 wt%-ZrH2 addition leads to the increase of the particle size of dispersoids and the decrease of the fracture strength and toughness.[6] On the other hand, doping ZrC can improve the mechanical properties of tungsten through refining grain size and inhibiting recrystallization.[1] Xie et al. reported that the trace nano-sized ZrC particles were added into W matrix by powder metallurgical process.[8] Both the ductility and strength are significantly improved in comparison with the commercial bulk pure W or W alloy, which can be attributed to coherent interface in grain/phase boundaries tuned by the purifying and strengthening abilities of the trace ZrC.[8] The first-principles calculations indicate that Zr element prefers to occupy the substitutional site in W with negative solution energy, suggesting that Zr element is easily dissolved in tungsten.[9–11] However, there have been few systematic studies of the properties for W–Zr alloy with different Zr concentrations and theoretical research is still required. The advent of density functional theory (DFT) has provided an alternative means of including the electron correlation to seek an insight into the mechanical mechanism.
Experimental results indicate that at room temperature and ambient pressure, W possesses a body-centered cubic (bcc) structure with a lattice constant of 3.165 Å (α phase). In the presence of oxygen, the W has a bcc structure with a lattice constant of 5.046 Å (β phase). However, the β phase transforms into the α phase when the temperature is higher than 630 °C and the process is irreversible. W as a plasma facing material withstand extremely high temperature. Thus, in this work, using the density functional theory, the effects of alloying Zr on the fundamental mechanical properties of W with α phase are studied. The structural, mechanical and electronic properties of W1−xZrx (x = 0.0625, 0.125, 0.1875, 0.25, 0.5) are calculated for different compositions. With the optimized geometry and lattice, some mechanical parameters such as bulk modulus (B), shear modulus (G), Youngʼs modulus (E), Poissonʼs ratio (υ), the B/G ratio, and Cauchy pressure (
2. Computational methods
First-principles calculations were performed with the pseudopotential plane-wave method implemented in the Vienna ab initio simulation package (VASP) code based on density functional theory (DFT).[12–14] The projector augment wave (PAW)[15] method was used to describe the core orbital, and the electron exchange correlation functional was calculated by using the Perdew–Burke–Ernzerhof approximation (PBE) described by the generalized gradient approximation (GGA).[16] The systematic calculations presented here have been performed on 2×2×2 supercell containing 16 atoms in a body centered cubic structure in W. Previous studies with first-principles calculations have shown that Zr atom can dissolve into bcc W to form substitutional binary solid solution.[17,18] In supercell calculations, the W alloying with different Zr concentrations, where W atoms are progressively replaced by Zr atoms have been studied. Six different Zr concentrations: no substitution (x = 0.0), one W atom replaced by one Zr atom (x = 0.0625), two W atoms replaced by two Zr atoms (x = 0.125), three W atoms replaced by three Zr atoms (x = 0.1875), four W atoms replaced by four Zr atoms (x = 0.25), eight W atoms replaced by eight Zr atoms (x = 0.5), have been performed for calculations. All possible configurations for each composition were here considered. The electron wave function was expanded in plane waves up to a cutoff energy of 350 eV. The calculated equilibrium lattice constant of pure W was 3.19 Å, and the result was consistent with the experimental value of 3.165 Å.[19] The Brillouin-zone integration was performed within Monkhorst–Pack scheme by using a 11×11×11 mesh for the geometry optimization and 13×13×13 mesh for electronic calculation.[20] For the case of pure Zr, the calculations have been performed on 2×2×2 supercell containing 16 atoms in a hexagonal close packing (hcp) structure and the 6×6×4 Monkhorst–Pack mesh has been used. Both supercell size and atomic positions were relaxed to equilibrium, and energy minimization continues until the forces on all atoms are converged to a value smaller than 10−2 eV/Å. The total energy of each system was relaxed until the difference value is smaller than 10−5 V.
We calculated the Cij components of the elastic tensor under the zero pressure from the computation of the stress–strain relationships.[21] According to Hookeʼs law, the stress–strain relationships can be described as

3. Results and discussion
3.1. Mechanical properties of bcc crystal of tungsten (W)
To check the accuracy of this computational setup, we calculate the single-crystal elastic constants and mechanical property parameters of pure W. The elastic constants and mechanical property parameters are given in Tables
| Table 1.
Elastic constants of bcc W. . |
| Table 2.
Bulk modulus (B), shear modulus (G), Youngʼs modulus (E), B/G ratio, Poissonʼs ratio (υ), Cauchy pressure |
3.2. Lattice constants and thermodynamic stability
The energy minimization procedure is used to obtain the total energy and optimal atomic relaxation geometries of all possible atomic configurations of W alloying with different Zr concentrations. The energy minimization shows that the W–Zr alloys can keep the bcc lattice and the most stable site for Zr atoms is located at the positions of the configurations in a high-symmetry. These results indicate that the W–Zr binary solid solution can be formed at an atomic level. The schematic diagrams of these high-symmetry configurations are illustrated in Fig.
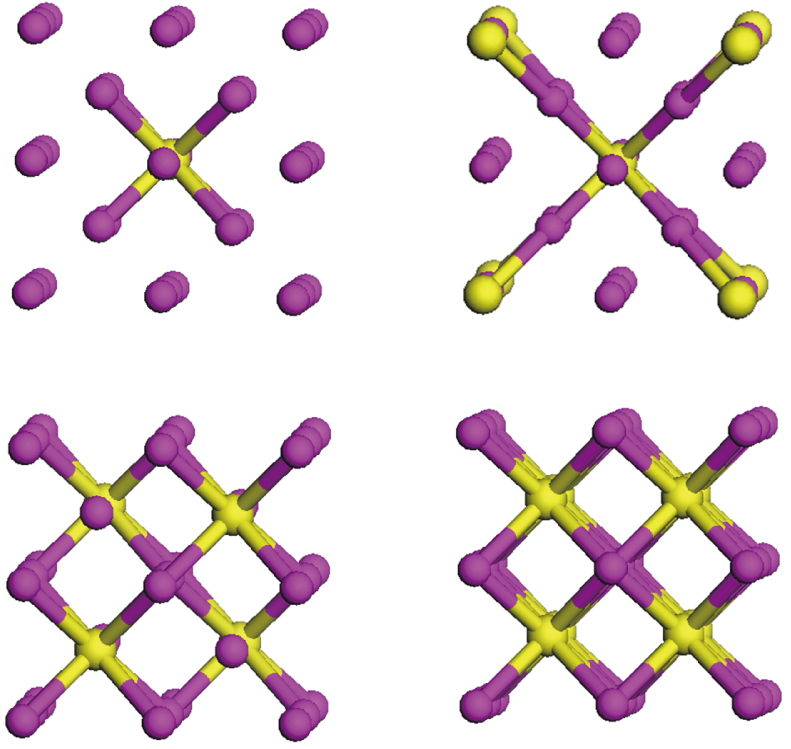 | Fig. 1. Schematic representation of energetically the most favorable atomic arrangements of the bcc W1−xZrx (x = 0.0625, 0.125, 0.25 and 0.5) in a 2×2×2 supercell. Small purple and large yellow balls represent W and Zr atoms, respectively. |
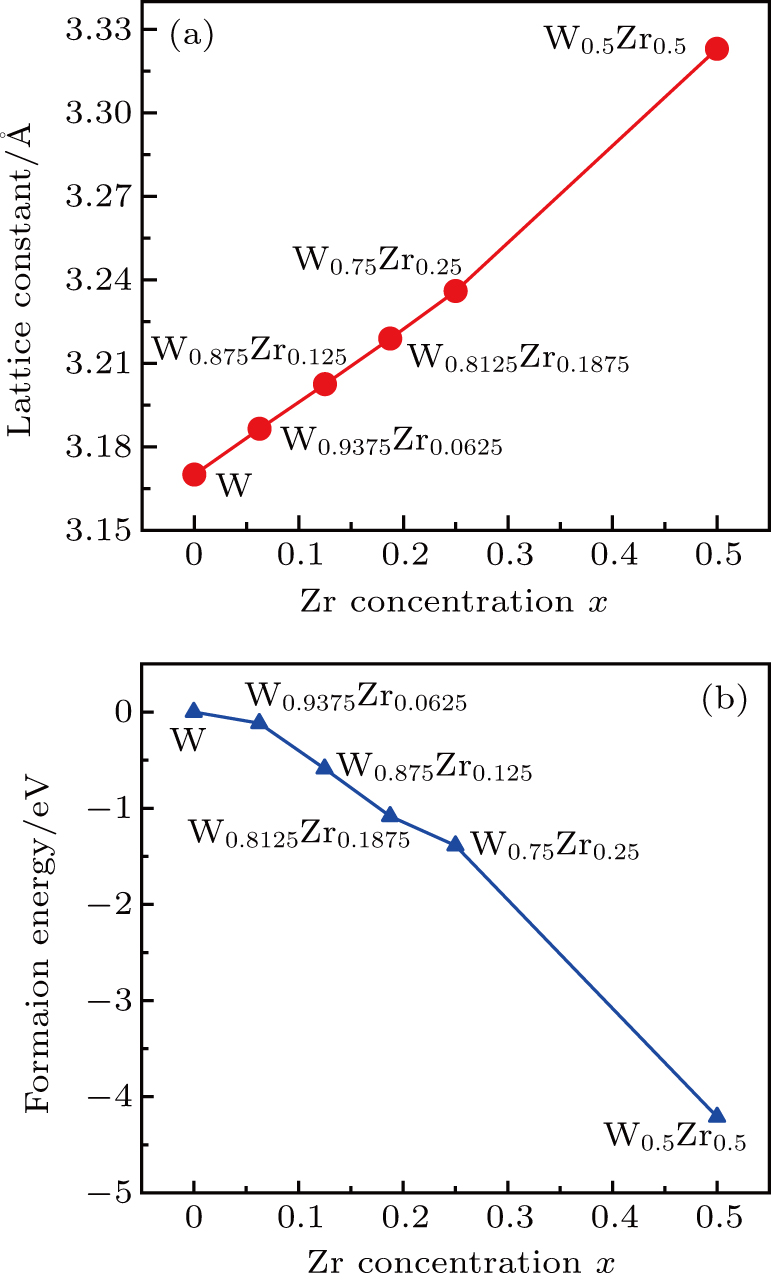
The lattice parameters of mixed crystals often obey the Vegardʼs law,[31,32] i.e., the lattice parameter of solid solution is linearly dependent on the concentration of one component and this is valid particularly for substitutional solid solutions.[33] Our calculations for the equilibrium lattice constants of W1−xZrx obtained from the total energy as a function of the volume are shown in Fig.
 | Fig. 2. Plots of (a) equilibrium lattice constants and (b) formation energy of bcc W alloying with Zr versus Zr concentration. |
A negative formation energy means that a structure is thermodynamically stable.[34–36] The effect of Zr concentration on the thermodynamic stability of W1−xZrx structure is evaluated from formation energy (Ef), which is defined as
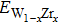
| Table 3.
Cell volumes, elastic constants, and formation energy of bcc W1−xZrx . |
3.3. Mechanical properties
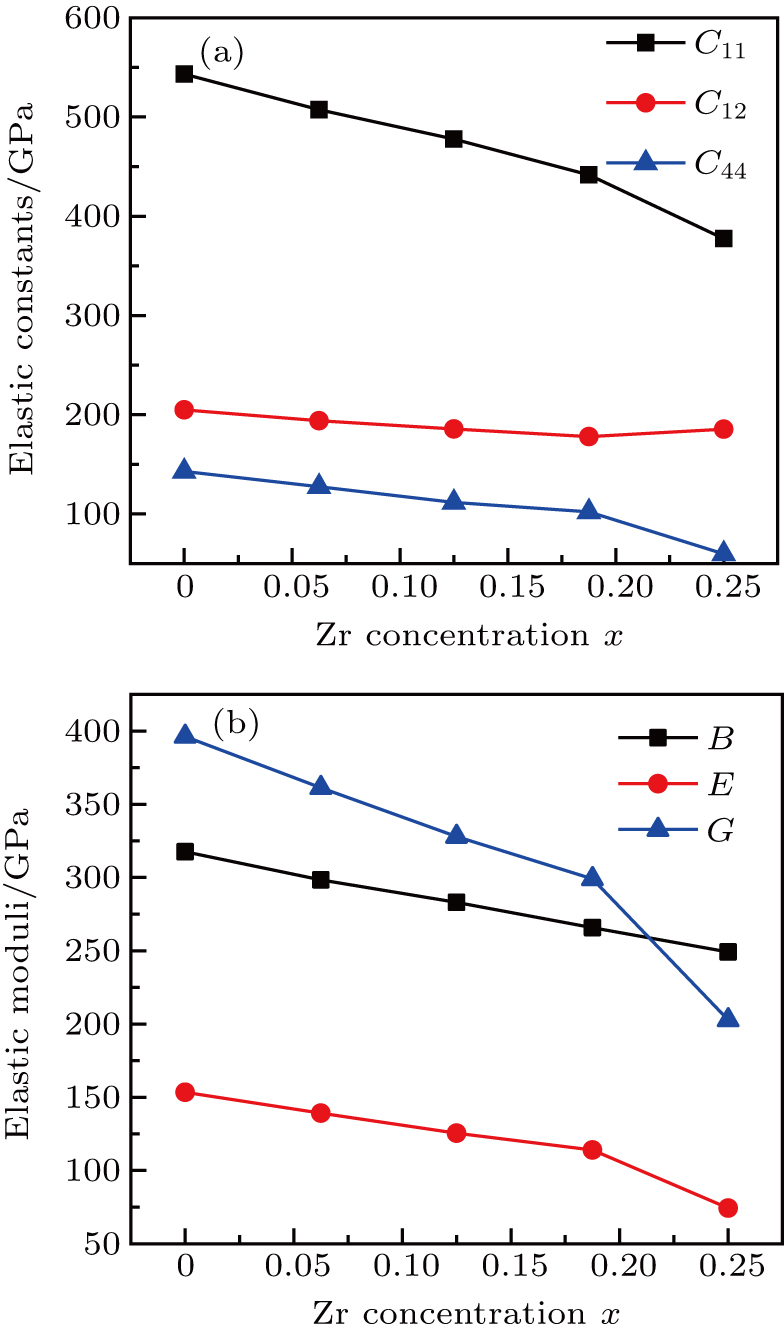
Now, we come to analyze the effect of an increased concentration of Zr on the elastic properties of the W1−xZrx solid solution. The calculated elastic constants relative to those of the pure W as a function of concentration x are given in Table
 | Fig. 3. (a) Elastic constants of bcc W alloying with Zr varying with Zr concentration; (b) Bulk modulus (B), shear modulus (G), and Youngʼs modulus (E) of bcc W alloying with Zr varying with Zr concentration. |
Our first-principles calculations explicitly indicate that the calculated elastic constants fulfill the above elastic stability criteria. The elastic stability of bcc W1−xZrx solid solution is guaranteed, thus ensuring a mechanically stable model of binary alloy systems. Interestingly, the C11 and C44 are clearly observed to decrease systematically (almost linearly), and the C12 varies with x slightly and nonlinearly. Indeed, the values of C11 reflects the stiffness of cubic crystal. The C11 value changes from 543.28 GPa for pure W to 245.98 GPa for W0.5Zr0.5, demonstrating that the stiffness decreases with the Zr content increasing by 6.25 at.%, until it reaches 50 at.%. In other words, the main elastic constants of the system decrease with Zr concentration, which makes the system softer.
To further understand the macroscopic mechanical properties of W1−xZrx, the values of derived bulk modulus (B), shear modulus (G), Youngʼs modulus (E), the ratio B/G, Poissonʼs ratio (υ), and Cauchy pressure (
| Table 4.
Values of bulk modulus (B), shear modulus (G), Youngʼs modulus (E), B/G ratio, Poissonʼs ratio (υ), Cauchy pressure |
The ratio of the bulk over shear modulus, B/G, has been proposed by Pugh[37] to identify a brittle or ductile behavior of the material. According to the Pugh criterion, a high B/G value (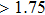
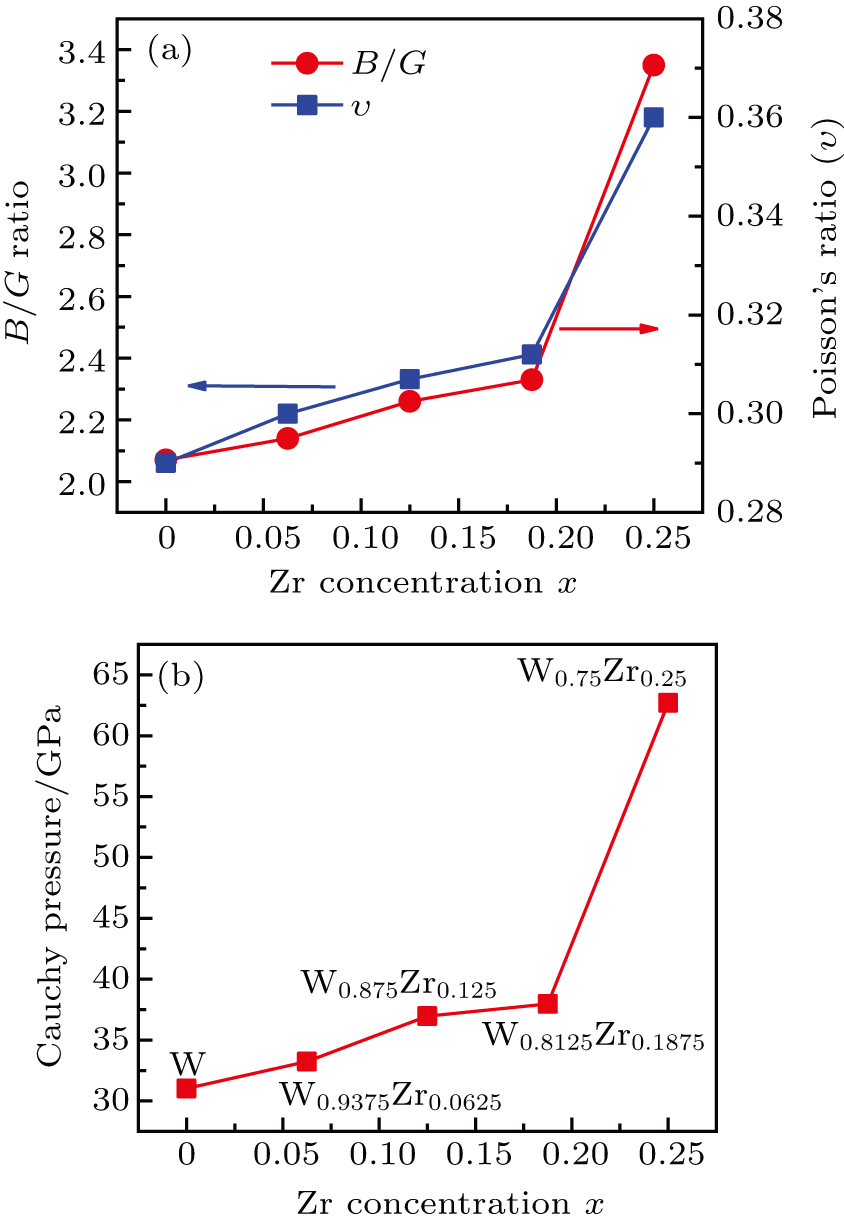 | Fig. 4. Plots of (a) B/G ratio and Poissonʼs ratio and (b) Cauchy pressure (
|
According to the analyses in the relevant research papers, the intermetallic compound of W2Zr is formed very easily when the temperature reaches a value between 1400 °C and 2210 °C, which will lead to the brittleness damage to W based materials. The intermetallic compound of W2Zr phase has a body-centered cubic structure and the phase prototype is W. Thus, the ductility of W2Zr phase should be improved based on our first-principles calculation. Noting that the data obtained by using first-principles simulation have limitations and in particular the effect of temperature on the data needs to be carefully assessed. Thus, the effects of temperature on W–Zr alloy and the properties of intermetallic compound of W2Zr will be studied in our future work.
The angular characteristic of atomic bonding, which also relates to the brittle/ductile properties of metals and metallic compounds, could be described by the Cauchy pressure.[42] For directional bonding with angular characteristic, the Cauchy pressure is typically negative. Meanwhile, for metallic bonding, the Cauchy pressure is positive, with larger positive pressure representing a more metallic characteristic and the better ductility. As shown in Table
3.4. Electronic structures
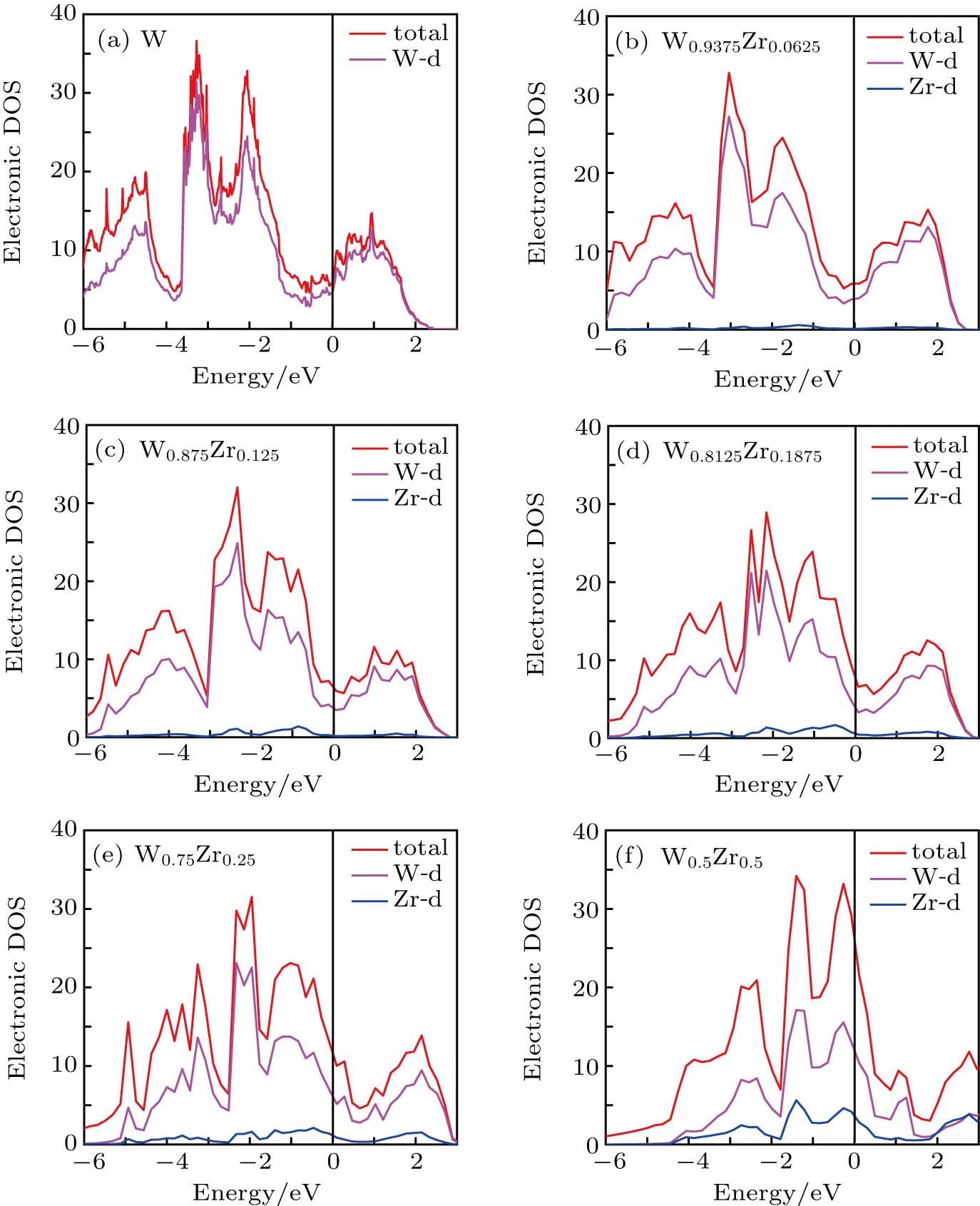
To understand the mechanical properties of W–Zr alloys, their underlying electronic structures are analyzed. Figure
 | Fig. 5. Density of states (DOS) of bcc-based chemically random W1−xZrx: (a) W when x = 1, (b) W0.9375Zr0.0625, (c) W0.875Zr0.125, (d) W0.8125Zr0.1875, (e) W0.75Zr0.25, (f) W0.5Zr0.5, with energy being relative to Fermi level. |
4. Conclusions
The structural, mechanical and electronic properties of W1−xZrx (x = 0.0625, 0.125, 0.1875, 0.25, 0.5) binary bcc substitutional alloys are systematically investigated by means of first-principles calculations within the framework of DFT. The formation energy values of all the studies W1−xZrx alloys are negative, indicating that they are all energy thermodynamically stable. Based on the calculations of the elastic constants, all the bcc W1−xZrx structures satisfy the mechanical stability criteria, i.e., the W–Zr binary substitutional solid solution can be formed at an atom level. Moreover, the elastic constants and the elastic moduli of W–Zr alloys are found to decrease gradually with the increase of Zr concentration, implying that the W–Zr alloys with higher Zr concentration become softer than pure W metal, although it is more rigid than the pure Zr metal. Meanwhile, by analyzing the value of B/G ratio, Poissonʼs ratio υ and Cauchy pressure 
Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] | |
| [33] | |
| [34] | |
| [35] | |
| [36] | |
| [37] | |
| [38] | |
| [39] | |
| [40] | |
| [41] | |
| [42] | |
| [43] | |
| [44] | |
| [45] |