

























































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Low-lying electronic states of aluminum monoiodide
Cite this Article
Yuan Xiang, Yin Shuang, Lian Yi, Yan Pei-Yuan, Xu Hai-Feng, Yan Bing. Low-lying electronic states of aluminum monoiodide. Chinese Physics B, 2019, 28(4): 043101 
Permissions
Low-lying electronic states of aluminum monoiodide
† Corresponding author. E-mail:
Abstract
Abstract
High-level ab initio calculations of aluminum monoiodide (AlI) molecule are performed by utilizing the multi-reference configuration interaction plus Davidson correction (MRCI+Q) method. The core-valence correlation (CV) and spin–orbit coupling (SOC) effect are considered. The adiabatic potential energy curves (PECs) of a total of 13 Λ–S states and 24 Ω states are computed. The spectroscopic constants of bound states are determined, which are in accordance with the results of the available experimental and theoretical studies. The interactions between the Λ–S states are analyzed with the aid of the spin–orbit matrix elements. Finally, the transition properties including transition dipole moment (TDM), Frank–Condon factors (FCF) and radiative lifetime are obtained based on the computed PEC. Our study sheds light on the electronic structure and spectroscopy of low-lying electronic states of the AlI molecule.
Keyword:AlI molecule;potential energy curves (PECs);core-valence correlation;spin-orbit coupling;multi-reference configuration interaction (MRCI)
1. Introduction
Aluminum monohalide plays an important role in the astrophysics[1] and molecular laser cooling.[2–6] The spectroscopic constants and the transition properties are critical to the understanding of the chemical and physical processes of these molecules. In recent years, aluminum monohalides have attracted increasing research interest due to their potential applications in molecular laser cooling. Based on the computed potential energy curves (PECs) and the spectroscopic constants, the laser cooling schemes using either AlF[6] or AlCl[4] and AlH,[6] have been suggested theoretically. As for AlBr,[5] however, the feasibility of laser cooling is still under debate,[4,5] owing to the fact that for heavy-atom-contained molecules, the electronic structure and spectroscopic constants are strongly affected with the inclusion of the core–valence correlation (CV) correction and spin–orbit coupling (SOC) effect.
While the spectrum and structure of electronic states have received considerable attention in previous experimental and theoretical investigations for lighter aluminum monohalides (AlF, AlCl, AlBr), our knowledge about AlI is still insufficient, especially for the electronic excited states, due to the abundant inner-shell electrons. To date, the experimental spectroscopic information is only available for the ground electronic state and the first triplet state.[7–9] Theoretically, Varga et al.[10] computed the molecular constants of the ground state by couple cluster single and double method combined with a perturbative treatment of triple excitation (CCSD(T)) method. The latest ab initio computation was performed by Hamade et al.[11] in 2009, in which the PECs and spectroscopic constants of 12 Λ–S states were investigated by using the multi-reference configuration interaction (MRCI) with single and double excitations plus Davidson correction (+Q). To the best of our knowledge, the SOC effect and CV correlation have not been investigated for AlI molecule. It has been indicated that these effects have significant influence on the electronic structures of the diatomic molecules containing heavy atom, such as PbO, ZnH and GeO.[12–14] Furthermore, the SOC effect leads to perturbations for the intercrossing of electronic states, changing the shape of PECs by the avoided crossing phenomenon. As a result, the spectroscopic and dynamic properties of electronic excited states are affected significantly due to the SOC effect.
The goal of this work is to achieve more accurate and detailed electronic structure of AlI molecule. A high-level ab initio calculation method using MRCI+Q is employed to compute a total of 13 singlet/triplet Λ–S states correlating with the lowest two dissociation limits of AlI molecule. The CV correction is considered via singly and doubly electron excitation from inner shells, and the SOC interactions between the Λ–S states are evaluated by the SO matrix elements. The 13 Λ–S states split into 24 Ω states when the SOC is introduced. The spectroscopic constants of low-lying Λ–S and Ω electronic states are obtained based on the computed PECs. We also acquire the transition properties of low-lying states including transition dipole moments (TDM), Frank–Condon factors (FCFs), and radiative lifetimes.
2. Computational details
The ab initio calculations of the electronic structure of the AlI molecule are performed with the quantum chemistry MOLPRO 2012 program package.[15] The 







We calculate 47 single point energies corresponding to the internuclear distance range of 1.9 Å–6.5 Å to obtain the adiabatic PECs of the ground and excited states. In these calculations, the Gaussian-type basis set cc-pwCVQZ-PP[16] in combination with corresponding relativistic effective core potential and cc-pwCVQZ[17] are selected for the I atom and the Al atom, respectively. The process of the calculation contains three steps. First, Hartree–Fock (HF) method is used for obtaining the single-configuration wavefunction of the ground state for the AlI molecule. Then, the state-averaged complete active space self-consistent field (SACASSCF)[18,19] is adopted. In the SACASSCF calculation, we involve a total of 13 electronic states including three 



The spin–orbit coupling effect is handled at the MRCI level. The spin–orbit coupling matrix element is dealt with the help of the state interaction approach via Breit–Pauli operator[22] for Al atom and ECP spin–orbit operator implemented in the ECP of I atom. The off-diagonal spin–orbit matrix elements are calculated at the MRCI level, however the diagonal spin–orbit matrix elements are substituted with MRCI+Q energies. The 13 Λ–S states split into 24 Ω states when we consider the influence of the spin–orbit coupling. The PECs of these 13 Λ–S states and 24 Ω states are depicted following the avoided crossing rule.
According to the PECs of the Λ–S and Ω states, the corresponding spectroscopic constants of the bound states are determined by resolving the one-dimensional nuclear Schrödinger equation with the aid of LEVEL program,[23] including excitation energy Te, equilibrium internuclear distance Re, vibrational constants ωe, and 
3. Result and discussion
3.1. PECs of 13 Λ–S states
The 13 Λ–S states of the AlI molecule, including seven singlet states and six triplet states, are studied by using the MRCI+Q method. Except for 3 




 | Fig. 1. PECs of Λ–S states for AlI molecule. |
As shown in this figure, the 



| Table 1.
Spectroscopic constants of bound and quasi-bound Λ–S states. . |
So far, only the spectroscopic constants of X 














| Table 2.
Main electric configurations of 1 |
Figure 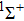
















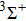
 | Fig. 2. Dipole moments of five Λ–S states. |
3.2. SOC effect on the AlI molecule
For the heavy molecule AlI, the effect of the SOC may not be neglected. The SOC effect causes the Λ–S states to split and mix with common Ω symmetry. From Fig. 




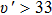
To analyze the interaction between the 13Π state and other states in detail, the SO matrix elements as listed in Table 
 | Fig. 3. R dependent SO matrix elements of several Λ–S states. |
| Table 3.
SO matrix elements of Λ–S state. . |
3.3. PECs of 24 Ω states
As discussed earlier, the SOC effect plays an important role in AlI molecule. After considering the SOC effect, the 13 Λ–S states split into 24 Ω states, including six states of 













 | Fig. 4. PECs of a total of 24 Ω states. |
 | Fig. 5. Curves of Λ–S component versus R of X0+, (1) 0−, (2)0+, (1)1, (1)2, and (2)1 states. |
| Table 4.
Dissociation limit relationships of Ω states. . |
| Table 5.
Spectroscopic constants of several bound Ω states. . |
The ground Ω state X+ almost completely originates from the Λ–S state X 







For the Ω states in the higher energy region, complicated state-mixing and interactions are presented under the influence of the SOC effect. Their PECs are very complex with many avoided crossing points. The (4)0+ and (5)0+ state hold the avoided crossing point around R=3.6 Å. Turing to the (5)1 and (6)1 state, there is an avoided crossing point at about R=3.3 Å. This generates a small potential well of (6)1 state. The state Ω=3 is purely generated from the 3Δ state, therefore, the shape of the PEC for Ω=3 is similar to that of the corresponding Λ–S state. Most of Ω states have shallow potential wells, but the depths of these wells are still too shallow to support many vibration levels and their Re values deviate significantly from that of the X0+ state. So, it can be expected that the spectra of the AlI in this energy range will be very diffuse and these states are hard to observe experimentally.
3.4. TDMs, FCFs, and radiative lifetimes of AlI molecule
where

is the degeneracy of the upper state,

is the Frank–Condon factor of the two vibrations levels, the energy difference

is in unit of cm−1, and TDM (in atomic unit) is the average electronic transition dipole moment. The results of the radiative lifetimes are listed in Table 7 . The computed radiative lifetimes of (2)0+ and (1)1 are

and

, respectively.
The transition dipole moments containing (2)1–X0+, (1)1–X0+, and (2)0+–X0+ are calculated each as a function of R in range from 1.9 Å to 6.5 Å. The corresponding functions of TDMs are depicted in Fig. 



 | Fig. 6. Transition dipole moments of AlI molecule. |
The Frank–Condon factors of the (1)1–X0+ and (2)0+–X0+ are calculated with the aid of LEVEL program as listed in Table 
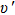
 |
| Table 6.
Franck–Condon factors of transitions (2)0+–X0+ and (1)1–X0+. . |
| Table 7.
Radiative lifetimes of the transitions (2)0+–X0+ and (1)1–X0+. . |
4. Conclusions
The ab initio calculations of the AlI molecule have been accomplished with the MRCI+Q method. The PECs of the 13 Λ–S states are calculated, which are related to the two dissociation limits Al(




Acknowledgment
The authors thank the High Performance Computing Center of Jilin University for computing time.
Reference
| 1 | |
| 2 | |
| 3 | |
| 4 | |
| 5 | |
| 6 | |
| 7 | |
| 8 | |
| 9 | |
| 10 | |
| 11 | |
| 12 | |
| 13 | |
| 14 | |
| 15 | |
| 16 | |
| 17 | |
| 18 | |
| 19 | |
| 20 | |
| 21 | |
| 22 | |
| 23 | |
| 24 | |
| 25 | |
| 26 | |
| 27 |