





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Hydrogenated antimonene as quantum spin Hall insulator: A first-principles study
Cite this Article
He Xin, Li Ji-Biao. Hydrogenated antimonene as quantum spin Hall insulator: A first-principles study. Chinese Physics B, 2019, 28(3): 037301 
Permissions
Hydrogenated antimonene as quantum spin Hall insulator: A first-principles study
† Corresponding author. E-mail:
Project supported by Research Funds of Sichuan University of Arts and Science, China (Grant No. 2012Z009Y).
Abstract
Using first-principles calculations based on density functional theory (DFT), the structural and electronic properties of hydrogenated antimonene have been systematically investigated. Phonon dispersion and molecular dynamics (MD) simulation reveal that fully hydrogenated (FH) antimonene has high dynamic stability and could be synthesized. A new σ-type Dirac cone related to b-px,y orbitals is found in FH antimonene, which is robust to tensile strain. Noticeably, the spin orbital coupling (SOC) opens a quantum spin Hall (QSH) gap of 425 meV at the Dirac cone, sufficiently large for practical applications at room temperature. Semi-hydrogenated antimonene is a non-magnetic metal. Our results show that FH antimonene may have great potential applications in next generation high-performance devices.
Keyword:antimonene;Dirac cone;quantum spin Hall (QSH) insulator;hydrogenated;first-principles calculations
1. Introduction
Since graphene was first isolated through mechanical exfoliation from ghaphite sheets in 2004,[1] two-dimensional (2D) materials such as transition metal dichacogenides, silicene, germanene, stananene, and phosphenene have been extensively studied[2,3] over the past decade. Owing to the unique honeycomb structure, 2D materials exhibit extraordinary properties, including topological insulator effect,[4] superconductivity,[5] and thermoelectricity,[6] which make them attractive for a wide range of applications in spintronics, optoelectronics, chemical sensors, and energy conversion or storage.[2,7,8] Recently, two novel graphene-like 2D materials, namely asenene and antimonene, were predicted through first-principles calculations by Zhang et al.[9] As the analogues of black phosphenene, asenene and antimonene have wide band gaps and high stability. To achieve multifunctional devices, it is crucial to tune the structural, electronic, and magnetic properties of 2D materials by various methods, including strain,[10] doping,[11,12] chemical functionalization,[13] and external electric field,[14] among which the chemical functionalization is an extremely effective way because of its high chemical reactivity.
Up to now, it has been reported that fully hydrogenated arsenene is a Dirac material and semi-hydrogenated arsenene exhibits significant magnetism.[15] Moreover, fully halogenated arsenene[16] and chemically decorated arsenene, AsX (X = CN, NC, NCO, NCS, and NCSe),[17] were also found to be Dirac materials. However, as far as we know, functionalized antimonene has not been reported till now. In this work, we investigate the structural and electronic properties of fully hydrogenated (FH) antimonene and semi-hydrogenated (SH) antimonene using first-principles calculations.
2. Method
First-principles calculations are performed within the framework of density functional theory (DFT) implemented in Quantum ESPRESSO package[18,19] using a norm-conserving pseudopotentials[20] and fully-relativistic ultrasoft pseudopotentials[21] for calculations without spin–orbital coupling (SOC) calculations and with SOC calculations, respectively. Spin polarization is included throughout the calculation. The exchange–correlation functional is treated using generalized-gradient approximation (GGA) of Perdew–Burke–Eruzerhof (PBE).[22] The unit cell optimization is terminated upon reaching the pressure cut-off of 0.01 kbar. The energy cutoff of the plane waves is set to 150 Ry in all computations except molecular dynamics (MD) simulation and nanoribbon. The energy criterion is set to 10−10 Ry for both electronic self consistency and structural optimization. A 20 Å vacuum layer is used to simulate the isolated sheet. The Brillouin zone integration is represented by the Monkhorst–Pack k-point scheme[23] with 15 × 15 × 1 grid mesh. All the lattice constants and atom coordinates are optimized until the convergence of the force on each atom is less than 10−5 Ry/bohr. The SOC is included in the self-consistent calculations of electronic structure. Phonon dispersion is obtained by lattice dynamic calculations performed using density functional perturbation theory[24] within linear response approach and by using the small displacement method[25] for antimonene and hydrogenated antimonene, respectively. First-principles MD simulation is performed with a canonical (NVT) ensemble. The simulation time step is 1 fs and the simulation time is 1 ps. The 4×4 supercell containing 64 atoms is equilibrated at 300 K. The energy cutoff of the plane waves is set to 40 Ry in MD simulation and for nanoribbon.
3. Results and discussion
Figure
 | Fig. 1. Top view (left) and side view (right) of optimized (a) pristine antimonene, (b) fully hydrogenated antimonene, and (c) semi-hydrogenated antimonene. |
| Table 1. Structural parameters calculated for pristine antimonene, fully hydrogenated antimonene, and semi-hydrogenated antimonene. Lattice constants a, buckling height Δ, bond angle θ, Sb–Sb bond length lSb−Sb, and Sb–H bond length lSb−H are given. . |
To verify the structural stability of pristine antimonene and hydrogenated antimonene, we perform phonon dispersion calculations. Figures
 | Fig. 2. Phonon band dispersions of (a) pristine antimonene and (b) FH antimonene. (c) Snapshot of FH antimonene structure in MD simulation at 300 K after 1 ps. |
Antimonene is a semiconductor with an indirect band gap of 1.24 eV in our calculations. The band structure with and without SOC and the corresponding total density of states (TDOS) and projected density of states (PDOS) of FH and SH antimonene are presented in Fig.
 | Fig. 3. Band structure with and without SOC and the corresponding TDOS and PDOS of (a) FH and (b) SH antimonene. |
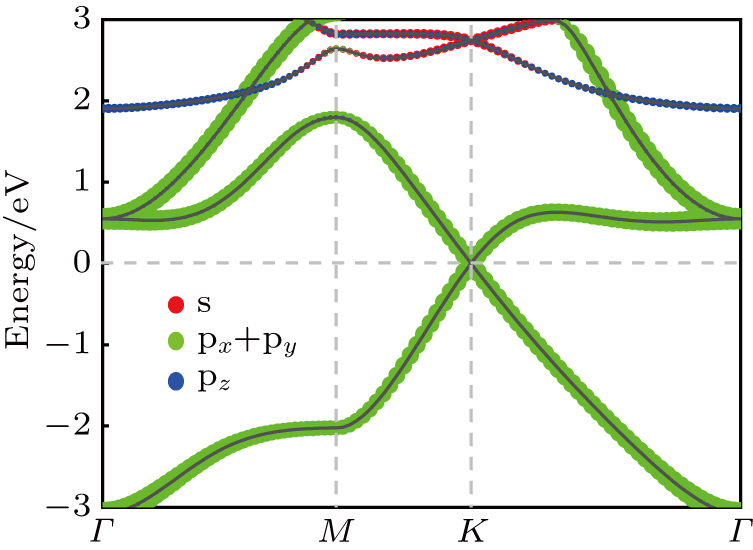 | Fig. 4. Projected band structure of FH antimonene with contribution from s, px+py, and pz orbitals represented by red, green, and blue dots, respectively. |
One of the prominent features of QSH insulators is the existence of gapless edge states that are helical with the spin-momentum locked. In order to explicitly demonstrate the edge states for FH antimonene, a zigzag nanoribbon (ZNR) with space inversion symmetry is constructed with the edges passivated by hydrogen atoms, as shown in Fig.
 | Fig. 5. (a) Top view of zigzag nanoribbon of FH antimonene with width of N = 7 (3.24 nm). (b) Band structure of ZNR for FH antimonene with (red) and without (blue) SOC. (c) The LDOS plots of ZNR. (d) Charge density distribuion in real space of the edge state near Fermi level. |
To see what would happen to Dirac cone in FH antimonene when the in-plane tensile strain is applied, we investigate the structural and electronic properties of FH antimonene under a biaxial strain. Here, a biaxial strain is defined as ε = (a−a0)/a0, where a and a0 are the lattice constants of the strained and the equilibrium unit, respectively. The band structure of FH antimonene with and without SOC under different strains are shown in Fig.
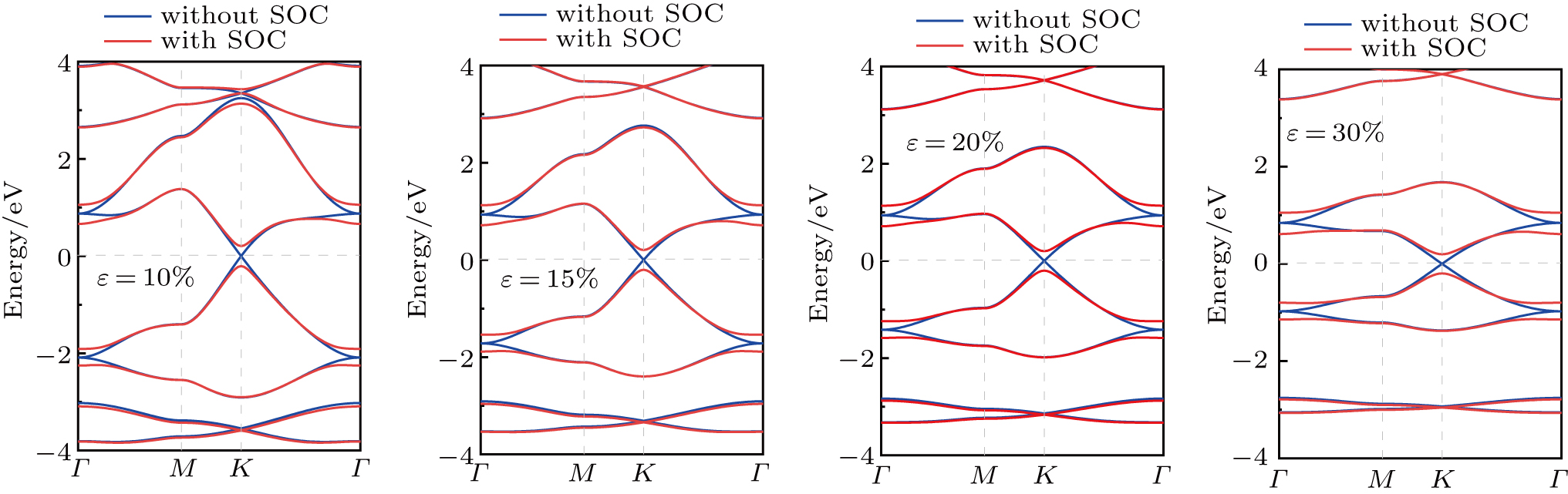 | Fig. 6. Band structure of FH antimonene without (blue) and with (red) SOC under different biaxial strain. |
4. Conclusion
In summary, we have explored the hydrogenated antimonene by first-principles calculations. FH antimonene is a QSH insulator with a band gap of 425 meV. Moreover, FH antimonene has high dynamic stability even at room temperature, indicating that it could be obtained in experiments. We have revealed that Dirac cone in FH antimonene is related to Sb-px,y orbitals, which is robust to tensile strain. SH antimonene is found to be a non-magnetic metal. We expect the experimental realization of FH antimonene and its great potential in next-generation high-performance devices.
Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] | |
| [33] | |
| [34] |