Chang Jing, Jiang Zhen-Yi, Song Qi, Chen Lei, Lin Yan-Min, Zhou Bo. Theoretical study on order–disorder phase transition of CH3NH3PbCl3*
Project supported by the National Natural Science Foundation of China (Grant Nos. 51572219, 51872227, 11204239, and 11447030), the Project of Natural Science Foundation of Shaanxi Province of China (Grant Nos. 2015JM1018, 2013JQ1018, 15JK1759, and 15JK1714), and the Science Foundation of Northwest University of China (Grant No. 12NW06).
. Chinese Physics B, 2019, 28(11): 116105
Permissions
Theoretical study on order–disorder phase transition of CH3NH3PbCl3*
Project supported by the National Natural Science Foundation of China (Grant Nos. 51572219, 51872227, 11204239, and 11447030), the Project of Natural Science Foundation of Shaanxi Province of China (Grant Nos. 2015JM1018, 2013JQ1018, 15JK1759, and 15JK1714), and the Science Foundation of Northwest University of China (Grant No. 12NW06).
Chang Jing1, Jiang Zhen-Yi1, 2, †, Song Qi1, Chen Lei1, Lin Yan-Min1, Zhou Bo1
Shaanxi Key Laboratory for Theoretical Physics Frontiers, Institute of Modern Physics, Northwest University, Xi’an 710069, China
Multidisciplinary Materials Research Center, Frontier Institute of Science and Technology, Xi’an Jiaotong University, Xi’an 710054, China
Project supported by the National Natural Science Foundation of China (Grant Nos. 51572219, 51872227, 11204239, and 11447030), the Project of Natural Science Foundation of Shaanxi Province of China (Grant Nos. 2015JM1018, 2013JQ1018, 15JK1759, and 15JK1714), and the Science Foundation of Northwest University of China (Grant No. 12NW06).
Abstract
Order–disorder phase transitions for CH3NH3PbCl3 are studied with density functional theory. Our calculations show that the disorder is manifested in two aspects in the cubic phase, namely, the disorder of orientation and rotation of organic groups. Organic groups of [CH3] and [NH3] in cubic crystals can easily rotate around its C3 axis. At the same time, [CH3NH3]+ organic groups can also orient to different spatial directions due to the weak interactions between organic group and inorganic frame. Our results show that its possible phase transition path starts from the deviation of organic groups from the crystal c-axis. Its structural transition changes from disordered cubic phase to hydrogen-only disordered tetragonal structure in the process of decreasing symmetry. The disordered high temperature cubic phase can be expressed as a statistical average of substructures we rebuilt. The electrostatic repulsive force between adjacent organic groups triggers out the formation of low temperature phase on cooling.
Organic-inorganic perovskite (ABX3), as a promising direct band gap semiconductor material,[1] has potential applications in solar cells due to its suitable band gap,[2] broadband spectrum,[3] long distance carrier transport,[4] low exciton binding energy,[5] large dielectric constant,[6] and absorption coefficient.[7,8] A large number of studies have focused on the properties of ABX3 materials. However, the study of phase transition is limited. Since the properties of crystals change due to phase transition, it is meaningful to understand the phase transformation process of crystals.
ABX3 series perovskite photovoltaic materials can be composed of a variety of A, B, and C, where A is usually methylammonium [CH3NH3]+,[9,10]B is a metallic element including Pb, Ge,[11,12] or Sn,[13]C is a halogen. CH3NH3PbCl3 can pass through three phases on cooling. CH3NH3PbCl3 holds cubic phase above 179 K and forms tetragonal structure at temperatures around from 172 K to 179 K.[14] The hydrogen atoms in [NH3] and [CH3] are located at the vertex of their groups, almost in the same plane of their respective groups, and are detected to exist in disordered states for cubic phase.[25] Furthermore, some uncertainties about the orientation of organic groups in CH3NH3PbI3 have also detected in the previous experimental observations,[15,16] which is the beginning behavior of order–disorder phase transition. The combination of organic groups with cubic octahedral cages of PbI6 plays a key role in their transformation process,[17] which has been confirmed by x-ray diffraction.[14] However, up to date no theoretical and experimental studies have been performed to understand these order–disorder phase transition behaviors. Inspired by the 12-site model proposed by Reynhardt and Hoon[18,19] we designed possible phase transition paths and obtained lower rotational barriers of organic groups, which will greatly improve the understanding of the mechanism of such phase transition.
2. Computational details
The ab initio calculations were performed in our work. Based on the density functional theory (DFT) of the VASP software,[20,21] we adopted the corrected projection plane wave (PAW) method[22,23] to describe the electron–electron and electron–ion interactions. A plane-wave cutoff energy is 500 eV in cubic phase and 450 eV in tetragonal phase. Tested 5 × 5 × 5 and 4 × 4 × 5 Monkhorst–Pack grids have been chosen for sampling of the Brillouin zone of cubic and tetragonal phases, respectively, showing an excellent convergence for total energy. Self-consistency was reached under a tolerance in total energy of 0.001 meV. The generalized gradient approximation (GGA) of Perdew–Burke–Ernzerhof (PBE) exchange correlation function[24] was applied in geometric optimization. In addition, the Tkatchenko–Scheffler method (TS) of van der Waals correction along the atom migration paths was used to accurately describe [CH3NH3]+ organic group and PbCl6 octahedral grid ion between inter-atomic forces. The time-consuming spin–orbit coupling method and the GW approximation method cannot describe the long-range electrostatic interaction better among organic groups, so these methods are abandoned in our calculations, where G refers to Green's function and W denotes the dynamically screened Coulomb interaction potential. In this paper, we abstract an ideal model based on classical electrostatic field theory to estimate the electrostatic energy between electric dipoles.
3. Results and discussion
3.1. The disordered state of cubic structure
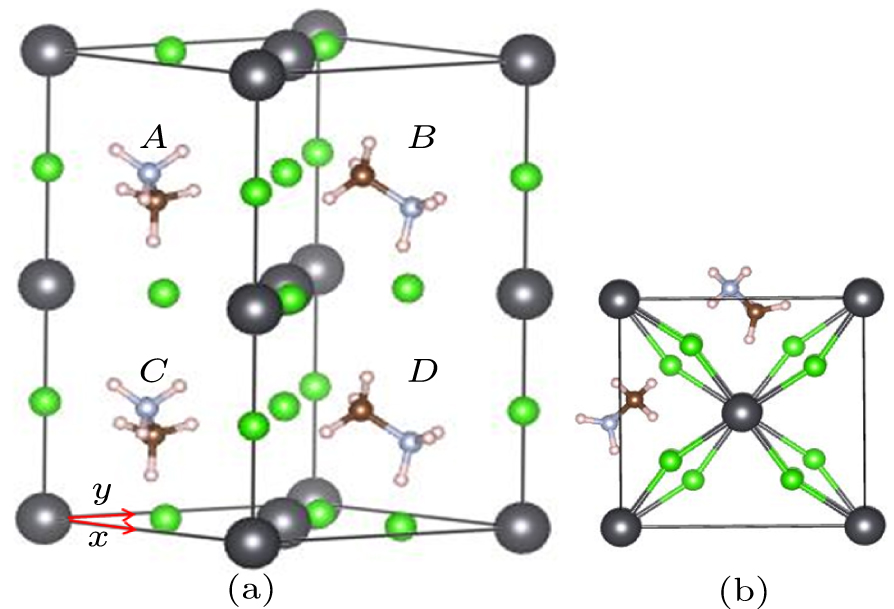
The order–disorder transformation has been found in the body-centered cubic structure[25] for CH3NH3PbCl3 (shown in Fig. 1) with symmetry above 179 K. There exists a centrosymmetric C3 axis (marked in Fig. 1) that overlaps C–N bond in this structure. For the purpose of better understanding the possible path of CH3NH3PbCl3 phase transition, we first discuss the disordered state of cubic structure.
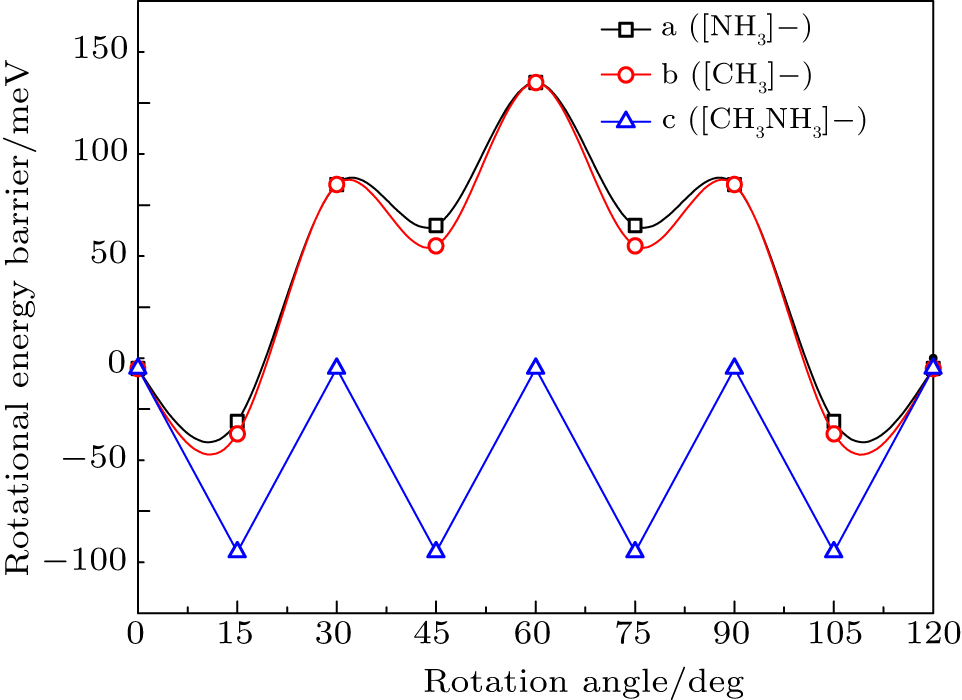
The disordered distribution of hydrogen atoms in cubic phase was detected in 1987.[25] Based on the 12-site instantaneous model theory previously used by Bindzus et al.,[18,26] similar models are also proposed here to study it in depth. Three hydrogen atoms in [CH3] or [NH3] groups were regarded as a regular triangle perpendicular to the C–N bonds and can rotate around centrosymmetric C3 axis (marked in Fig. 1). Their rotational energy barriers of three hydrogen atoms from staggered to eclipsed conformations is shown in Fig. 2. The highest barrier is about 142 meV and is slightly smaller than the average kinetic energy (about 182 meV) of [CH3] or [NH3], which means that hydrogen atoms can rotate freely around the C3 axis at the same level, and only the statistical average structure can be determined. The repulsive electrostatic energy between the three H atoms in [CH3] and the three H atoms in [NH3] increases by about 100–200 meV during the rotation of [CH3] around the C3 axis (as Fig. 1) from 0° to 60°. Considering that the highest rotational potential barrier of the hydrogen atom is only about 142 meV, it is obvious that repulsive electrostatic energy plays a key role in the disorder of hydrogen atoms.
Fig. 2. Rotatioal energy barriers of [CH3]/[NH3]/[CH3NH3] group around the C3 axis in cubic phase.
Series of fractional-occupancy configurations were designed by rotation of the orientation axis of [CH3NH3]+ along different directions. According to the rotation operation and the corresponding energy, these substructures are divided into four groups, An, Bn, Cn, and Dn series. In series-A structures, the orientation axis of [CH3NH3]+ rotates 15° from [001] to [010] in the (200) plane. In series-D structures, the orientation axis of [CH3NH3]+ rotates 15° from [001] to [110] in the () plane. The rotations of the C and D series take place in the plane between (200) and (), and correspond to 15° and 30° in Fig. 1, respectively. Thus, 6 × 4 = 24 configurations are obtained from all of the rotations around the above A, B, C, and D axes (as shown in Fig. 3). Our calculations show that their highest barrier among 24 configurations is about 226 meV, which corresponds to the path from A2 to A3 (as shown in Fig. 4). Most of them have barriers less than 150 meV, which is slightly smaller than the average kinetic energy (about 182 meV) of [CH3NH3]+ group. Therefore, most substructures between A, B, C, and D series can be converted to each other, especially between A1, A2, B1, B2, C1, and C2.
Fig. 4. Energy barrier of 24 space configurations (relative to original cubic structure) with hydrogen atoms in staggered state.
In addition, the disorder of hydrogen in [CH3] or [NH3] and the disorder of orientation of whole [CH3NH3]+ may exist simultaneously. During this rotation of [CH3NH3]+ orientation axis, total 24 × 5 = 120 possible structures were designed considering that three hydrogen atoms in [CH3] or [NH3] could rotate further around that orientation axis (5 conformations from staggered to eclipsed in Fig. 1) at the same time. Their single point energies of 120 possible configurations were calculated with the optimized cubic structure (a = b = c = 5.79 Å, and α = β = γ = 90°). The total energies of these configurations depend on organic group orientations and their disordered hydrogen. From the initial structure to the 120 substructures, the maximum potential barrier is about 425 meV. The structure with lower energy is located in the region of the 24 substructures, in which the triangular hydrogen atoms of [CH3] and [NH3] groups lie in staggered states (as shown Fig. 1). Limited to space, there is no further illustration here.
3.2. Tetragonal structure
As shown in Fig. 5, the relaxed unit cell of CH3NH3PbCl3 is the tetragonal phase (I4/mcm) with lattice parameter a = 8.4 Å, b = 8.4 Å, c = 12.5 Å in the temperature range 173–179 K.[25] The tetragonal structure can be obtained from the cubic structure by rotation of 45° in-plane and double along c-direction, and then the lattice parameters of tetragonal structures are times in comparison to cubic structures.[5] Furthermore, the orientation of tetragonal organic group [CH3NH3]+ can be regarded as the deviation of organic group [CH3NH3]+ from [001] direction in cubic phase. Its Pb–Cl–Pb bond angles no longer keep straight but form an obtuse angle. The Pb–Cl–Pb bond angles change from about 180° to 160°, and the [CH3NH3]+ atoms in a staggered petal geometric shape are no longer perpendicular to the xoy plane, but form 30° angle instead. In addition, the organic groups A and B are away from each other in order to form a more stable spatial orientation with a minimum repulsive force.
Fig. 5. Tetragonal phase of CH3NH3PbCl3 (front view in (a) and overhead view in (b)).
3.3. Phase transition
The phase transition process is always accompanied with the change of the atomic motion and energy. It is believed that the phase transition is dominated by the motion of the [CH3NH3]+ group in the cubic phase. The atomic kinetic energy reduces with decreasing temperature, and the organic group in the crystal will gradually adopt those orientations with lower potential energy. The rotation motion of O–C1, O–B1, O–A1, C1–C2, B1–B2, A1–A2, C2–A2, and B2–A2 is frozen in turn and eventually most of them ended up in A2. Their barrier among various substructures with different orientations is low enough that it will not be too slow for the dynamic process from those to the energetically lowest orientation. The transition path from O to A1 and then to A2 is the energetically easiest in Fig. 4.
In order to understand the origin of transition driving force for [CH3NH3]+ group deviating from [001] lattice axis, we abstract [CH3NH3]+ into an electric dipole according to the calculated charge number. From the Bader charge calculation, [NH3] and [CH3] groups are seen as negative and positive charges with −0.272e and +0.5905e, respectively. The orientations of the adjacent electric dipoles in cubic phase are described by angles of θ and ϕ (θ = 0°–180°, ϕ = 0°–180°). Their dipole interaction energies are shown in Fig. 6, where the lowest energy point is located at F. From the dipole-parallel state to the F-point state, the electrostatic energy is reduced by about 120 meV. By comparing with the angle between adjacent dipoles in the tetragonal phase, it is found that they have the same intersecting solid angle (about 150°). Therefore, the driving force of phase transition from cubic to tetragonal phase mainly comes from the interaction between electric dipole. As the temperature decreases, the eight adjacent organic groups in the pseudo-tetragonal crystal unit tend to F-structure (about 150°) with each other, which leads to symmetry breakdown and forming a tetragonal phase structure. Certainly, the reorientation of eight organic groups triggered the reduction of Pb–Cl–Pb bond angles, which could further reduce the lattice distortion energy of the system.
Fig. 6. Potential energy differences of dipole interaction with different orientations, comparing with the parallel dipole.
4. Conclusion
We have investigated the phase transformation path of CH3NH3PbCl3 from disordered cubic to tetragonal structure. High temperature cubic phase is disordered and mainly manifested in two types: the disorder of orientation of organic groups and the disorder of hydrogen atoms. Hydrogen atoms can randomly rotate around the C3 axis at high temperatures, whereas the [CH3] and [NH3] prefer to be staggered. The high temperature ordered structure obtained from the experimental observations is actually the average of various disordered structures. The original symmetry of cubic phase breaks on cooling, and the [CH3NH3]+ group can easily convert among the orientations of O, A1, A2, B1, B2, C1, and C2 states. With the decreasing temperature, the rotation motion of O–C1, O–B1, O–A1, C1–C2, B1–B2, A1–A2, C2–A2, and B2–A2 is frozen in turn and eventually most of them ended up in A2. The dipole interaction of [CH3NH3]+ groups in adjacent cubic unit cell promotes the formation of their cross re-orientation. Eventually, periodic cubic CH3NH3PbCl3 cells are re-chosen and the tetragonal phase with the I4/mcm symmetry is obtained.
Theoretical study on order–disorder phase transition of CH3NH3PbCl3*
Project supported by the National Natural Science Foundation of China (Grant Nos. 51572219, 51872227, 11204239, and 11447030), the Project of Natural Science Foundation of Shaanxi Province of China (Grant Nos. 2015JM1018, 2013JQ1018, 15JK1759, and 15JK1714), and the Science Foundation of Northwest University of China (Grant No. 12NW06).
[Chang Jing1, Jiang Zhen-Yi1, 2, †, Song Qi1, Chen Lei1, Lin Yan-Min1, Zhou Bo1]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}