











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Ethylene glycol solution-induced DNA conformational transitions
Cite this Article
Zhang Nan, Li Ming-Ru, Zhang Feng-Shou. Ethylene glycol solution-induced DNA conformational transitions. Chinese Physics B, 2018, 27(11): 113102 
Permissions
Ethylene glycol solution-induced DNA conformational transitions
† Corresponding author. E-mail:
Project supported by the National Natural Science Foundation of China (Grant Nos. 11635003, 11025524, and 11161130520), the National Basic Research Program of China (Grant No. 2010CB832903), and the European Commissions 7th Framework Programme (FP7-PEOPLE-2010-IRSES) (Grant No. 269131).
Abstract
We study the properties of the ethylene glycol solutions and the conformational transitions of DNA segment in the ethylene glycol solutions by molecular dynamics simulations based on GROMACS. The hydrogen network structures of water–water and ethylene glycol–water are reinforced by ethylene glycol molecules when the concentrations of the solutions increase from 0% to 80%. As illustrated by the results, conformation of the double-stranded DNA in ethylene glycol solutions, although more compact, is similar to the structure of DNA in the aqueous solutions. In contrast, the DNA structure is an A–B hybrid state in the ethanol/water mixed solution. A fraying of terminal base-pairs is observed for the terminal cytosine. The ethylene glycol molecules preferentially form a ring structure around the phosphate groups to influence DNA conformation by hydrogen interactions, while water molecules tend to reside in the grooves. The repulsion between the negatively charged phosphate groups is screened by ethylene glycol molecules, preventing the repulsion from unwinding and extending the helix and thus making the conformation of DNA more compact.
1. Introduction
Water is the most important life supporting solvent.[1,2] Two thirds of the environment surrounding the DNA molecules in the cell nucleus is composed of water molecules.[3,4] This provides an appropriate environment for both biophysical and biochemical processes[5–7] to develop. A basic question of interest is which fundamental property of its molecule distinguishes water as the most important solvent in nature.[8,9] Are there any other liquids that support biological activity? An important property of water is that it dissolves charged ions and polar molecules easily. It is generally considered that polarity and hydrogen bonding are the distinguishing attributes of water when compared with other liquid.[10,11]
Many experimental and theoretical studies have reported[12–18] the properties of DNA macro-molecule in neutral aqueous. There are many kinds of solvents, such as acids or alkaline solvents, organic or inorganic solvents, and hydrophilic or hydrophobic solvents. It is ostensibly undesirable that strong acids and strong alkaline solutions are corrosive, or DNA is insoluble in most organic solvents. An exception is the organic solvent ethylene glycol. It has been observed that DNA can retain its native double-stranded structure in ethylene glycol–salt solutions.[19,20] The biological activity of DNA in ethylene glycol has been investigated by following the transforming activity of bacillus subtilis DNA with competent bacteria.[21] DNA can keep its biological activity after long time periods in ethylene glycol–salt solutions. However, DNA in ethylene glycol has been little investigated during last half century.
In addition, ethylene glycol solutions have the following advantages in comparison with aqueous solutions.
(i) It is known that radiation damage ionizes the water molecule and creates hydroxyl radicals[22–25] which adversely affect a cell's DNA duplexes. It seems possible that DNA might be more resistant to radiation damage in ethylene glycol than in water, since hydroxyl radicals would not be generated in similar amounts in irradiated ethylene glycol solutions. The randomly distributed clusters of diatomic OH-radicals that are the primary products of megavoltage ionizing radiation in water-based systems are the main source of hydrogen-abstraction as well as formation of carbonyl and hydroxyl groups in the sugar moiety that create holes in the sugar rings. These holes grow slowly between DNA bases and DNA backbone with the damage affecting both DNA single and double strand breaks.[26,27] Various effects of the ionizing radiation on biological systems range from the development of genetic aberrations, carcinogenesis to aging.[28,29] Ethylene glycol may provide better protection for DNA against cosmic radiation, hence facilitating its safe interplanetary transport.
(ii) It is possible to work with double-stranded DNA in ethylene glycol solutions in the convenient temperature range of −10 °C to +25 °C, where micro-organisms do not usually grow.[19] Thus the ethylene glycol may present a cheaper and more efficient option for storing DNA in bio-banks than in frozen aqueous buffer solutions.
Computational modeling can help to gain a detailed understanding of DNA structure and function. The most accurate approach would encompass the use of the quantum mechanical (QM) wave function. However, realistic modeling of DNA molecule and its environment requires extensive computing resources and can only be applied to a limited number of conformations. Therefore, most simulation projects employ classical molecular mechanics (MM) energy functions. Because of the need for high statistical computation of these functions during the simulation, the force fields used are relatively simple and utilize many adjustable empirical parameters which are generated by fitting the experimental properties or quantum chemical calculations.
Theoretically, the properties of ethylene glycol solutions and the conformational transitions of DNA are all determined by the microscopic interactions between the solvents and DNA molecules. In various experimental methods (x-ray crystallography, neutron diffraction, nuclear magnetic resonance (NMR) and fluorescent techniques, and atomic force microscope) and theoretical methods, the method of the molecular dynamics (MD) simulation can help us understand many macroscopic and microscopic phenomena of inter- and intra-molecular interactions, and simultaneously pave a useful way of discovering flexibility and tracing conformation transitions at atomic level[30–35] which would complement the static structure imaging techniques and help interpret experimental results in structure studies. Using MD simulation method with different parameters, we can study the effect of ethylene glycol on the hydrodynamic properties of DNA in solution. Their results are helpful for understanding the properties of DNA in ethylene glycol solutions with applications in radiation protection and DNA store.
In this paper, a series of water and ethylene glycol mixture solvents with five different weight concentrations of ethylene glycol are designed by the PACKMOL code package.[36] Conformational transitions of the duplex d(CGCGAATTCGCG) in ethylene glycol are studied at 293 K temperature using MD simulation. As concentration rises, the hydrogen network between water–water and ethylene glycol–water changes. Ethylene glycol then forms hydrogen bond with the negatively charged oxygen atoms of phosphate groups around the backbone.
2. Model and methods
2.1. DNA and solvent models
where ε0 is the vacuum permittivity and rij presents the distance between two atoms. The Lennard–Jones core provides short-range inter-molecule repulsion and describes the molecular size.[46] The Coulomb interactions between point charges describe the effects of hydrogen bond and give the local orientational structure.[47]
The Drew–Dickerson dodecamer (DDD) has been chosen for the initial state of DNA (PDB ID: 171d). The reason for this choice is that (i) DDD contains the recognition site of the EcoRI restriction enzyme and is frequently used in gene recombination techniques,[37] and (ii) it has been extensively studied experimentally and in nanosecond-to-microsecond MD simulations.[38–40] In addition, the DNA double strand is long enough to maintain a complete B-helix so that we can make the model more representative of a longer double-stranded DNA sequence. Stereo views of the initial 171d are given in Fig.
 | Fig. 1. (color online) Stereo views of the initial 171d structure. Presented here are 3 typical views from major groove, minor groove, and top. The systems are treated with the PARMBCS1 force field. |
Although some sophisticated potential functions of water are still being developed,[8] in this work the most widely used TIP3P model is adopted because its description is, generally, satisfactory under ambient conditions when compared with experiments.[41] PARMBSC1 force fields are used to describe oligonucleotide interactions in ethylene glycol solutions (Table
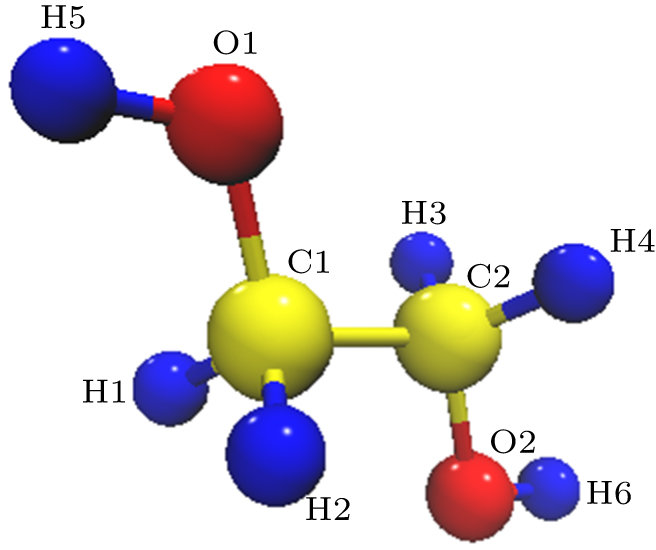 | Fig. 2. (color online) Stereo views of ethylene glycol. The red, yellow, and blue represent oxygen, carbon, and hydrogen atom, respectively. |
| Table 1. The force field parameter for ethylene glycol models in molecular dynamics simulations. . |
The intermolecular energy consists of two parts: E = ELJ + EC, where ELJ is the Lennard–Jones interaction and EC is the electrostatic interaction. They are expressed as
 |
 |
2.2. Simulation description
Classical MD simulations have been performed based on GROMACS code packages to study the conformational transitions of DNA-molecule in the ethylene glycol solution.[48] One DNA dodecamer, 22 Na+ counter-ions, and 8040 solvent molecules (water and ethylene glycol, big enough to ensure that the DNA does not interact with its periodic images) are contained in a periodic cubic box. The aim of this work is to study the solvent effects on DNA conformation. Two groups of parallel simulations are conducted.
The first one is the simulation of the behavior of aqueous ethylene glycol mixtures at 293 K. The data cover the entire range of composition at 20% mass intervals. The time step is 2 fs and the total simulation time is 200 ns. A complete overview of the simulation systems is given in Table
After that, 171d segment has been put into the concentration of solvents 40%, with 22 Na+ counterions for each system. The time step is 2 fs and the total simulation time is 200 ns.
Each of these molecular dynamics simulations consists of four stages. At the beginning, we employ the energy minimization to ensure that the system has no steric clashes or inappropriate geometry. Then the system is thermalized and pre-equilibrated using our standard multi-step protocol[50] followed by 1 ns of equilibration. Next, an isothermal–isobaric ensemble (P = 1 atm and T = 293 K) uses the Berendsen algorithm[51] to control the temperature and the pressure, with a coupling constant of 5 ps. Finally, an MD production run of 200 ns is performed to provide configurations for analysis.
| Table 2. Overview of ethylene glycol/water simulations: number of molecules, ethylene glycol mass fraction Ci, mole fraction Xi, and fractional error δ(ρ). Experimental densities from Ref. [49] are the equilibrium properties of the mixtures. . |
2.3. DNA structure parameters
Structure parameters describing the axis-base pair (X-displacement, Y-displacement, inclination), intra-base pair (shear, stretch, stagger, buckle, propeller, opening), inter-base pair (rise, roll, shift, slide, tilt, twist), backbone torsion (BI/BII population, canonical α–γ, puckering), and groove characters (MW, mW, MD, mD) of DNA are analyzed by programs Curves+ and Canal.[52] The X-displacement (Xdisp) describes the displacement of a base pair along the X direction (the direction of the short axis of the base pair) of the base pair axis. Inclination (Incl) is defined as dihedral angle between the base pairs and the plane with the initial x and y axis. Twist and roll ρ are relative rotation around the z axis and rotation around the y axis between two successive base pairs in an overall helix axis, respectively.[53] The width (MW for major groove and mW for minor groove) and depth (MD for major groove and mD for minor groove) of the groove are defined in Ref. [53]. All of these parameters are analyzed continually for the precise values in our simulations. The B-DNA conformation can most easily be distinguished from the A conformation with these parameters.
The data for the DNA in pure water and ethanol solutions are from the BIGNASim database (http://mmb.irbbarcelona.org/BigNASim/index.php).
3 Results and discussion
3.1. Properties of ethylene glycol solutions
Firstly, we demonstrate the information correctly on the molecular interactions between water and ethylene glycol in binary mixtures at a series of weight percent of ethylene glycol. The calculated results are compared with the experimental data. For easy comparison, the simulated densities are tabulated in Table
In the mixtures solvent of 20% (Table
Figure
 | Fig. 3. (color online) The radial distribution functions of water and ethylene glycol in a series of concentrations glycol solutions. (a) water oxygen (ow) –ow, (b) ow–water hydrogen (hw), (c) hw–hw, (d) ethylene glycol oxygen (o1) –ow, (e) o1–hw, and (f) o1–o1. Mass fractions 0%, 20%, 40%, 60%, and 80% are shown as orange, black, red, green, and blue, respectively. |
Results for ethylene glycol–water RDFs are shown in Figs.
The behavior exhibited by the first peaks may be explained by the spatial density functions (SDFs) of water around one ethylene glycol molecule as indicated in Fig.
 | Fig. 4. (color online) The spatial density functions of water around one ethylene glycol molecule in (a) 20%, (b) 40%, (c) 60%, and (d) 80% ethylene glycol solvents. The scale of the isosurface is the relative density of water. The red, yellow, and blue represent oxygen, carbon, and hydrogen atom, respectively. |
The first peak of g(r)o1–o1 occurs at 0.28 nm, similar to that exhibited in water–water g(r)ow–ow, revealing that the ethylene glycol–ethylene glycol hydrogen bonding persists, even as this component becomes more diluted. An examination of Figs.
The changes of hydrogen-bonded network structure in these solutions can be presented in Fig.
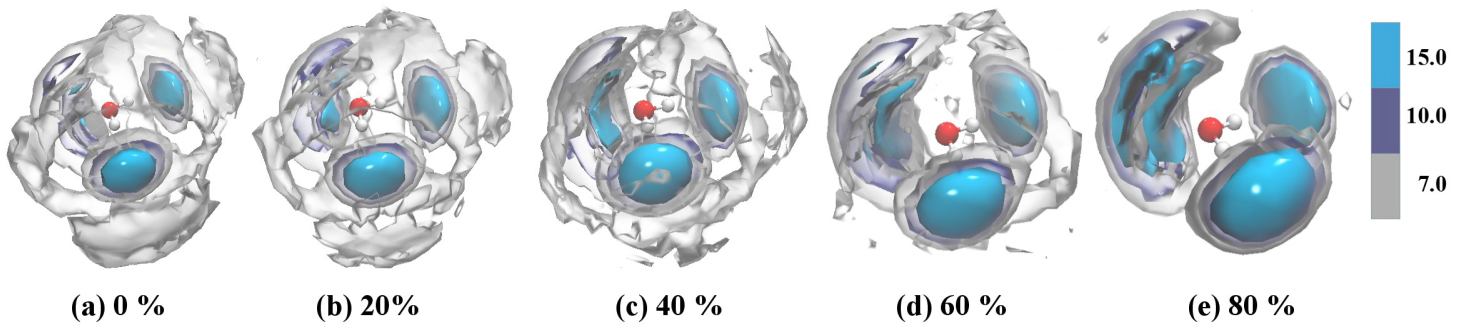 | Fig. 5. (color online) The spatial density functions of water around one water molecule in (a) 0%, (b) 20%, (c) 40%, (d) 60%, and (e) 80% ethylene glycol solvents. The scale of the isosurface is the relative density of water. The red and blue represent oxygen and hydrogen atom, respectively. |
Comparing effects of different concentrations shows that the ρr = 15 and 10 isosurfaces occupy large areas in high concentrations solutions. This is particularly noticeable for the nearest neighbors compared to the central oxygen site. As the concentration of the solutions increases, the second hydrogen shell of water is destroyed. This effect is seen in Fig.
The information on the molecular interactions between water and ethylene glycol at a series of concentrations is important for our understanding of complex interactions between DNA and ethylene glycol solutions. Here we present the change of H-bonded network structure induced by concentration change through some distributions functions such as radial distribution functions and spacial density functions. Firstly, revealing the presence of hydrogen bonding in water–water, ethylene glycol–water, and ethylene glycol–ethylene glycol, the intermolecular hydrogen bonding combinations have the following priority: water–water ˃ ethylene glycol–water ˃ ethylene glycol–ethylene glycol. As the hydrogen bonded network structure is affected by the concentration, the first shell layer between water–water and ethylene glycol–water molecules increase. On the other hand, the distribution of the first shell molecules between the ethylene glycol–ethylene glycol molecules is not obviously affected by the concentrations of the ethylene glycol solutions.
3.2. Conformational transitions of DNA
It was earlier shown that the transforming activity of DNA from bacillus subtilis was unaffected in salt-containing ethylene glycol solutions at room temperature. We now discuss the analysis of the 171d in ethylene glycol solutions. Molecular dynamics simulations were first carried out at 293 K to access the thermodynamic stability of the DNA structures in ethylene glycol solutions.
To study the thermodynamic stability of DNA conformation, the duplex DNA is put into the TIP3P model solvent with ethylene glycol (Ci = 40) at 293 K. At Ci = 20, it seems that the main structure features are similar to those observed for pure water. However, as the weight percent of ethylene glycol increases, the average error for the densities also increased. Thus the 40% weight percent of ethylene glycol is chosen as the solvent environment for the simulations of DNA.
The main results are presented in Fig.
 | Fig. 6. (color online) (a) RMSD of the DNA trajectories with respect to the initial structure (black line), x-ray (red line), and NMR (blue line). (b) Evolution of the total number of Watson–Crick hydrogen bonds with time. Red solid is the ideal hydrogen bond number between the 12 base pairs while the red dashed line is the ideal hydrogen bond number without the terminal base pair. (c) Evolution of the total number of hydrogen bonds between water and DNA with time. (d) Evolution of the total number of hydrogen bonds between ethylene glycol and DNA with time. (e) Evolution of the total number of hydrogen bonds between ion and DNA with time. (f) Evolution of the total number of hydrogen bonds between solvents (water+ethylene glycol+ions) and DNA with time. The running averages over 1 ns are shown in blue line. The weight percent of ethylene glycol solution is 40%. |
In general, we observe that DNA can retain a stable structure in ethylene glycol solutions. A more detailed discussion of DNA conformational transitions is carried out below. We begin with a discussion of the impact on DNA structures by water/ethylene glycol solvents. We then turn to a discussion of the average structural parameters and a comparison with other solutions.
3.2.1. Solvent impacts on DNA
We first look at the evolution of the total number of Watson–Crick hydrogen bonds with time, shown in Fig.
In this paper, the hydrogen bonds are analyzed through two different programs, GROMACS and VMD. It is worth noting that the hydrogen bond determination criteria in different programs may not be consistent. In fact, there are multiple criteria for the determination of hydrogen bonds, such as energy criteria, electronic structure criteria, geometric criteria, and so on. The first two criteria are computationally intensive and can not be achieved with classical molecular dynamics simulations. Therefore, the geometric criteria is used in this manuscript. Even geometric criteria still have different forms. In this paper, the hydrogen bond is determined by the cut-off of the angle formed by the hydrogen donor–acceptor and the cut-off of the donor–acceptor distance. The standard for determining the presence of H-bonds is the distance between the acceptor and donor atom ˂ 3.5 nm (corresponding to the first minimum position of the SPC water model RDF) and angle between the acceptor and donor atom ˂ 30°.[54] The blue indicates the averages at 1 ns intervals.
Also shown are: (i) in the range 0–50 ns, the number of hydrogen bonds exceeding the ideal number, (ii) the number of hydrogen bonds being in the range 32–26 majority of time, and (iii) in the last 15 ns, the number of hydrogen bonds being higher than the ideal value. The number of hydrogen bonds in aqueous solutions are rarely found to be higher than the ideal value. Double-stranded DNA reversibly attains[55] a more compact and less extended conformation with a smaller solvent shell in ethylene glycol than in water. This structural feature may provide the possibility of more hydrogen bonds forming in the Watson–Crick strand.
To illustrate the hydrogen bond characteristics of DNA in the glycol solution, we present several conformations in Fig.
 | Fig. 7. (color online) Representative structures of the characteristics hydrogen bonds conformations found. |
 | Fig. 8. (color online) Representative structures of the characteristic hydrogen bonds conformations found. |
Figures
These structural features are short-lived and have no significant effect on the overall structure of DNA. A long-lived conformation is shown in Fig.
The stepped shift, observed in the vicinity of 50 ns in Fig.
This dynamical feature is consistent with DNA in aqueous solution in previous MD simulations[38-40,56] and experiments.[57,58] However, in the case of aqueous solution, not only neither recent molecular dynamics simulations nor available experimental data support the presence of long-lived open state for terminal base pairs, but they reject the possibility of long-lived non-canonical conformations of terminal base pairs stabilized by interactions with the DNA grooves. We will discuss the reasons for this difference below.
Overall, there is a case for the number of hydrogen bonds exceeding the number of ideal hydrogen bonds for a long period which may be related to the more compact structure of the DNA in the ethylene glycol solution. A more compact structure may provide conducive conditions for the formation of hydrogen bonds in base pairs. We have found some short-lived non-canonical conformation, such as the phosphate acid bond break between the 3C and 4G and the sub state structure of 11C–14G base pair. We also found a long-lived non-canonical conformation, the fraying of the terminal base pair. Compared to their cytosine partner, no torsional flips are observed for the terminal guanine. The population of the non-canonical conformation is sequence-dependent, with lower probability on the central AATT.
We now look at the interaction of DNA molecules with solvents and counter-ions in solution. First of all, from Figs.
Figure
The discussion of hydrogen bonds is summarized as follows. (i) There is a case where the number of hydrogen bonds between base pairs in DNA exceeds the number of ideal hydrogen bonds numbers. (ii) A long-lived terminal damage occurs. (iii) Both ethylene glycol molecules and water molecules interact with DNA through hydrogen bonds and there is a competition between them. (iv) Ions interact with DNA through strong chemical bonds.
It has been suggested that the physical and chemical mechanism of these DNA helices are relevant to solvent accessibility, base stacking interactions, phosphate hydration, and minor groove spine of hydration.[59–61] The most commonly concerned factor should be how the repulsion between negatively charged phosphate groups is screened.[62] Because the repulsion will unwind and extend the helix without screen and the different screen mechanism also affects the pipeline of the electrostatic repulsion, to check the underlying cause of the structural transitions induced by the solvent, we evaluate the water and ethylene glycol distribution around the phosphate groups on DNA helix.
The SDFs of the ethylene glycol molecules around the DNA are presented in Fig.
 | Fig. 9. (color online) (a) The spatial density functions of ethylene glycol molecules around DNA in ethylene glycol solvents. (b) The spatial density functions of water molecules around DNA in ethylene glycol solvents. (c) The spatial density functions of Na+ around DNA in ethylene glycol solvents. |
An interesting phenomenon observed, in contrast to the DNA in aqueous solution, is a ring structure of ethylene glycol molecules around the phosphoric acid backbone. This is, as previously stated above, due to the number of hydrogen bonds being higher than the ideal number in ethylene glycol solutions. Analysis of the spatial distribution functions shows that the repulsion between negatively charged phosphate groups is screened by ethylene glycol molecules and extends the helix. Herskovits et al. observed[55] that double-stranded DNA reversibly attains a more compact and less extended conformation with a smaller solvent shell in ethylene glycol than in water, while retaining intact biological activity. Because of the structure of DNA compact, it provides more possibility for hydrogen bonds in Watson–Crick strand. It is possible that the distribution of terminal cap-shaped ethylene glycol molecules causes long-life terminal fray.
3.2.2. Structural parameters
The structural parameters analysis is divided into the sub-sections: (i) backbone torsion, (ii) axis base pair, (iii) intra-base pair helical parameters, (iv) inter-base pair helical parameters, and (v) grooves.
The three major elements of flexibility in the backbone are (a) rotations around ζ/ε torsion, (b) rotations around α/γ torsion, and (c) sugar puckering.
The concerted rotation around ζ/ε torsion generates two major conformers: BI and BII, which are experimentally known to co-exist in an approximate ratio of 80%:20% (BI:BII) in B-DNA.[63] Figures
 | Fig. 10. (color online) Population of BI/BII substate in (a) ethylene glycol, (b) ethanol, and (c) aqueous solutions. Population of canonical α/γ conformation in (d) ethylene glycol, (e) ethanol, and (f) aqueous solutions. |
A comparison of the characteristics in Figs.
Analysis shows that in ethylene glycol solution, the BII population is higher than 90% in 4G step, which is obviously higher than the other two solutions. The location of this peak corresponds to the breaking point of the chain between 3C–4G, as discussed earlier. The populations of BII in ethanol solution are significantly lower than those in other two solutions, and higher than 30% in the middle base 7T.
On the basis of a statistical analysis of x-ray data, Djuranovic and Hartmann discovered that passing from BI/BI to mixed BII/BII substates implies a decrease in roll and increase in twist.[65] The data reported by Heddi et al.[66] indicate that slide increases, too. This is expected to be particularly important in understanding indirect recognition of DNA by proteins,[67,68] and also the mechanism of DNA intercalation by small organic compounds.[69]
Another structural deviation common in simulations is the flip of the backbone torsion angles α/γ from the canonical g−/g+ state to the non-canonical state as shown in Figs.
In ethylene glycol solution, there is a non-canonical α/γ torsion at 2G step, the population of canonical g−/g+ state is 29%, the population of non-canonical g−/g− (only γ flips) is 35%, g+/g+ (only α flips) is 27%, t/g+ (only α flips) is 7%, and others is 2%. Flipping tends to occur in one of α/γ, while α/γ has a similar probability of flipping. The probability of α/γ flipping at the same time is only 2%. In ethylene glycol solution, the non-canonical α/γ flips with low probability at other bases. In ethanol solution, non-canonical α/γ flipping appears at 10% of the time, and the distribution is more dispersed in bases. The non-canonical distribution in aqueous solution is also very scattered, but the non-canonical state in pure water solution only appears 3%. The presence of isolated non-canonical α/γ conformers leads to a local decrease in the twist but has little impact in other helical parameters.[39] The unusual α/γ conformation could be a factor favoring recognition by specific proteins, given that the crystal structures of protein–DNA complexes show a significant percentage of such states.
The sugar puckering is divided into four states based on ranges of the sugar pucker phase angle: north puckering (315°–45°), east puckering (45°–135°), south puckering (35°–225°), and west puckering (225°–315°). In ideal B-DNA double helix, the sugar ring is C2'-endo folded belonging to south region. In ideal A-DNA double helix, the sugar ring is C3'-endo folded and belongs to north region. Figure
 | Fig. 11. (color online) Population of nucleotides with north puckering (pink), east puckering (blue), west puckering (yellow), and south puckering (green) in (a) ethylene glycol, (b) ethanol, and (c) aqueous solutions, respectively. |
As expected for water and ethylene glycol solutions, the sugar puckering is mostly in the south region, accounting for 82% and 70%, respectively. There is also an average of 16% conformation in the east puckering for the aqueous solution, and average of 27% conformation in the east–south mixing zone for the ethylene glycol solution. Among them, the south region (C2'-endo) belongs to the ideal B-DNA, implying that the DNA configuration is a mixture of type B and other configurations (east puckering) in water and glycol solutions. In contrast, the two conformations south and north puckering in the ethanol solution are in a dynamic equilibrium, accounting for 55% and 35%, respectively, and the north puckering is mainly in the middle 8 base pairs. The south to north transition produces a moderate increase in the width of the minor groove,[39] among which the north region (C3'-endo) belongs to the ideal A-DNA, which means that the DNA configuration is a mixture of type B and type A in ethanol solutions (10% east puckering).
Study of the DNA backbone torsion shows that the conformations of DNA in ethylene glycol and aqueous solutions are more similar, whereas those in ethanol/water mixtures are clearly different from each other. DNA is more prevalent in B-type conformations in ethylene glycol and aqueous solutions, whereas in ethanol/water mixtures, it is more prevalent in A–B mixed conformations.
To evaluate the DNA structure changes in detail, 8 distinct helical parameters, which are distinguishing between A and B forms, are calculated[52] and presented in Fig.
 | Fig. 12. (color online) Comparison of average values of helical parameters per base pair step from ethylene glycol, ethanol, and aqueous solutions. |
For the base pair helical axis parameters, the standard deviation at the 8T17A base pair is larger in ethanol solution. Table
| Table 3. The effect of backbone torsional changes on the helical parameters. . |
4. Conclusion
We have presented the properties of ethylene glycol solutions and the conformational transitions of DNA on the structure in ethylene glycol solutions by using a large scale molecular simulations.
We found that the hydrogen bond network structure changes induced by the concentration increase of the ethylene glycol solutions through different kind of distribution functions such as RDF and SDF. Both of these distribution functions give the picture that the number of molecules in the first shell layer between ethylene glycol–water and water–water molecules increased with the increase of the concentration of ethylene glycol solutions. On the other hand, the distribution of the first shell molecules between the ethylene glycol–ethylene glycol molecules is not obviously affected by the concentration of the ethylene glycol solutions. Furthermore, the intermolecular hydrogen bond interactions occur between water–water, ethylene glycol–water, and ethylene glycol–ethylene glycol, with the following order of priority: water–water ˃ ethylene glycol–water ˃ ethylene glycol–ethylene glycol.
Under the conditions of 293 K and 1 atm pressure, the conformations of 171d in ethylene glycol solutions are stable and no major distortions are found except for long-life terminal damage, which usually occurs on the terminal cytosine without appearing on the paired guanine. The cytosine undergoes a torsional flip to form a sub-state with a hydrogen-bond forming between the oxygen atom (on the sugar ring of the terminal cytosine) and the hydrogen atom (on the sugar ring of the 3G). In addition, the hydrogen atom (on the sugar ring of the terminal cytosine) formed hydrogen bond with the oxygen atom (on the sugar ring of the 4C).
Analysis of the total number of Watson–Crick hydrogen bonds in ethylene glycol solution shows occurrence of a situation where the number of the hydrogen bonds is higher than the ideal number (32 hydrogen bonds). It may be due to the double-stranded DNA reversibly attaining a more compact and less extended conformation with a smaller solvent shell in ethylene glycol than in water.
The double-stranded DNA in ethylene glycol solution were similar to the structure of DNA in the aqueous solutions but more compact. The reason may be that (i) ethylene glycol molecules and water molecules have the consistent interactional mechanism for the DNA (hydrogen bonds), such that the conformation of DNA in ethylene glycol solutions is similar to that in aqueous solution; (ii) ethylene glycol molecules have higher hydrogen bonding capacity than water molecules to bind to phosphate acid negative oxygen atoms in DNA. The ethylene glycol molecules preferentially distribute around the phosphate groups and minor groove to influence DNA conformation by hydrogen interactions, while water molecules prefer to reside in the grooves. Thus the negatively charged phosphate groups are screened (the repulsion will unwind and extend the helix). In addition, we also added the DNA structure in the ethanol/water mixed solution as a contrast and found that the DNA was in an A–B hybrid state in the ethanol/water mixed solution.
This paper presents a unified quantitative and qualitative method of describing the transitions of DNA conformation in ethylene glycol solutions based on molecular simulation. Although various relevant factors, such as the concentrations of solutions, pressure conditions, and species of ions, were investigated, further studies of these effects and their underlying physical mechanisms are suggested.
Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] | |
| [33] | |
| [34] | |
| [35] | |
| [36] | |
| [37] | |
| [38] | |
| [39] | |
| [40] | |
| [41] | |
| [42] | |
| [43] | |
| [44] | |
| [45] | |
| [46] | |
| [47] | |
| [48] | |
| [49] | |
| [50] | |
| [51] | |
| [52] | |
| [53] | |
| [54] | |
| [55] | |
| [56] | |
| [57] | |
| [58] | |
| [59] | |
| [60] | |
| [61] | |
| [62] | |
| [63] | |
| [64] | |
| [65] | |
| [66] | |
| [67] | |
| [68] | |
| [69] |