




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Charge compensation and capacity fading in LiCoO2 at high voltage investigated by soft x-ray absorption spectroscopy
Cite this Article
Long Xing-Hui, Wu Yan-Ru, Zhang Nian, Yu Peng-Fei, Feng Xue-Fei, Zheng Shun, Fu Jia-Min, Liu Xiao-Song, Liu Na, Wang Meng, Xu Lei-Min, Chen Jin-Ming, Lee Jenn-Min. Charge compensation and capacity fading in LiCoO2 at high voltage investigated by soft x-ray absorption spectroscopy. Chinese Physics B, 2018, 27(10): 107802 
Permissions
Charge compensation and capacity fading in LiCoO2 at high voltage investigated by soft x-ray absorption spectroscopy
† Corresponding author. E-mail:
Project supported by the National Natural Science Foundation of China (Grant Nos. 21503263, U1632269, 21473235, and 11227902).
Abstract
Abstract
In order to obtain an in-depth insight into the mechanism of charge compensation and capacity fading in LiCoO2, the evolution of electronic structure of LiCoO2 at different cutoff voltages and after different cycles are studied by soft x-ray absorption spectroscopy in total electron (TEY) and fluorescence (TFY) detection modes, which provide surface and bulk information, respectively. The spectra of Co L2,3-edge indicate that Co contributes to charge compensation below 4.4 V. Combining with the spectra of O K-edge, it manifests that only O contributes to electron compensation above 4.4 V with the formation of local O 2p holes both on the surface and in the bulk, where the surficial O evolves more remarkably. The evolution of the O 2p holes gives an explanation to the origin of 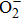
1. Introduction
Layered LiCoO2 cathode, which is used in the first commercial lithium ion battery (LIB), still dominates the portable electronic device market nowadays.[1,2] Its theoretical capacity can reach up to 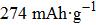
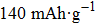
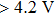
Because the lithiation/delithiation are always accompanied with insertion/extraction of electrons into/from the electrodes, investigation on the evolution of electronic structure of LiCoO2 will provide new information for better understanding of the origin of extra capacity at high voltage and the mechanism of capacity fading.[16–20] Goodenough pointed out that the Co4+/Co3+ redox couple at the top of the O band gave an intrinsic voltage limit and that the formation of peroxide with 
Several sXAS studies focusing on the electronic structure of LiCoO2 have been reported.[24–26,28–30] Montoro et al.[24,25] showed that the Co ions remain mostly unaffected and always in trivalent low-spin states with Li deintercalated chemically. On the other hand, the O was partially oxidized during Li chemical extraction. Uchimoto et al.[26] studied the electronic structure of LiCoO2 during the electrochemical reaction and concluded that the Co ions were still 3+ even with 0.8Li extracted from LiCoO2 while the oxidation of O ions mainly took place. Conversely, Yoon et al.[28] demonstrated that charge compensation could be achieved in O and Co ions simultaneously with LiCoO2 film electrode cycled in a range between 3 V and 4.2 V. They also suggested that the capacity fading of the LiCoO2 could be related to the decrease of Co–O bond covalency. Chen et al.[30] studied the chemically delithiated LiCoO2 and also demonstrated the contribution of Co ions should not be negligible in the charge compensation process. Meanwhile, they found that the oxidation of O2− on the surface and in the bulk were different. Mizokawa et al.[41] emphasized the critical role of O 2p holes and thought that the O 2p holes were bonded to the Co4+ sites and Li+ vacancy, which helped to improve the electronic and Li+ conductivities. Obviously, the charge compensation from O has been confirmed through the sXAS studies, which strongly supports the model proposed by Goodenough. Meanwhile, the contribution of Co ions to the charge compensation process has also been verified gradually. Nevertheless, the detailed mechanism of the charge compensation from O is still under debate. According to Goodenough’s model, LiCoO2 will suffer structure collapse due to the O loss at the voltage higher than 4.2 V. However, all the studies above did not give any further description of the evolution of O related to the O loss on the surface or in the bulk. It should also be noted that the evolution of O 2p holes needs to be studied carefully because of the convolution of features at the pre-edge of O K-edge sXAS resulting from O 2p hole and the hybridization between Co and O. Furthermore, the correlation between capacity fading and evolution of electronic structure during cycling with the cutoff voltage higher than 4.2 V invokes more investigation.
In this work, we study the LiCoO2 cathodes of commercial pouch cells from Contemporary Amperex Technology Ltd (CATL, Ningde, Fujian). Benefiting from the strict manufacturing processes, a solid evolution of surface and bulk electronic structure could be explored by measuring soft x-ray absorption spectra in TEY and TFY modes with surface (

2. Experimental methods
2.1. Battery test and sample preparation
Pouch full cells were prepared by the CATL Company. The cathode was composed of LiCoO2, super P black (SP), and polyvinylidene difluoride (PVDF) in a weight ratio of 94:3:3. The anode was made of hard carbon and PVDF binder in a weight ratio of 92:8. The electrolyte of LiPF6 (
All cells were tested by using the electrochemical workstation at 45 °C. For the 1st cycle, the cells produced in the same batch were firstly charged to 3.9 V at a rate of 0.1 C then to a certain cutoff voltage (i.e., 4.2 V, 4.4 V or 4.6 V) at a rate of 0.7 C and kept for 0.2 h, and then discharged to 3 V at a rate of 0.7 C and kept for 0.2 h. For the subsequent cycling test, the schedule was almost the same as that in the 1st cycle except that the charging rate to 3.9 V is 0.7 C.
All the tested cells were transported into Ar-filled glovebox and disassembled at an Ar atmosphere with O2 and H2O content lower than 0.1 ppm. The cathode samples were washed three times by using anhydrous DMC to remove the interference of electrolyte. To avoid any possible interference from the atmosphere, the samples were sealed with Al-plastic bags, taken to the Taiwan Light Source (TLS), and then transferred to the test chamber through an Ar-filled glovebox.
The samples with various cutoff voltage including 3.9 V, 4.2 V, 4.4 V, and 4.6 V at the 1st cycle were prepared for the charge compensation study. Samples at fully charged (4.4 V or 4.6 V) or discharged (3 V) states with the different capacity retention ratios (i.e., 100%, 99%, 51%, 42% at 4.6 V, and 100%, 72%, 50% at 3 V) and samples cycled 10 times between different voltage ranges (3 V–4.4 V versus 3 V–4.6 V) were prepared for studying the correlation between the evolution of the electronic structure and capacity fading.
2.2. Soft x-ray absorption spectroscopy
The sXAS measurements of the LiCoO2 cathode were carried out at beamline 20A1 of the Taiwan Light Source (TLS) at HsinChu, Taiwan, beamline 08U of Shanghai Synchrotron Facility (SSRF) and beamline 12B-a of the National Synchrotron Radiation Laboratory (NSRL) at Hefei. In particular, the beamline 20A1 was equipped with a 6-m high-energy spherical grating monochromator (6-m HSGM) to supply a photon beam with resolving power up to 8000. The spectra of Co L2,3 and O K-edge were collected in TEY and TFY mode in an ultrahigh-vacuum chamber with a base pressure of about 
3. Results and discussion
3.1. Competition between amount of Li+ extraction and cycling performance
The amount of Li+ extraction with various cutoff voltages is shown in Fig.
 | Fig. 1. (color online) Electrochemical tests of LiCoO2, showing (a) amount of Li+ increasing with upper cutoff voltage, and (b) cycling performances for the charge cutoff voltages of 4.4, 4.5, and 4.6 V. |
The cycling performances with the upper cutoff voltage of 4.4, 4.5, and 4.6 V are shown in Fig.
3.2. Charge compensation during Li extraction
To avoid any possible interference from the side reaction at the electrode/electrolyte interface, the TFY spectra of Co L2,3-edge and O K-edge, which reflect the bulk evolution, are firstly studied. As shown in Fig.
 | Fig. 2. (color online) (a) Normalized Co L2,3-edge x-ray absorption spectra and (b) normalized oxygen K-edge x-ray absorption spectra of Li1−xCoO2 with respect to the cut-off voltage in the TFY mode, and ((c) and (d)) magnified parts of Co L3 and O K pre-edge spectra above 4.4 V, respectively, which are convenient for comparison. |
When charged to 3.9 V, the intensity of peak B increases notably while the peak A shifts to higher energy and becomes broader. During charging from 3.9 V to 4.4 V, the intensity of peak B continues to increase while peak A continues to shift to higher energy and become broader. However, there is almost no change when charging from 4.4 V to 4.6 V, as confirmed by the careful comparison in Fig.
Next, the corresponding spectra of O K-edge are studied. As shown in Fig. 
What is more attractive is the evolution of O K-edge above 4.4 V where there is no obvious variation at the Co L2,3-edge. By careful comparison, as shown in Fig. 


3.3. Surficial evolution with Li extraction
The TEY spectra of Co L2,3 are shown in Fig.
 | Fig. 3. (color online) (a) Normalized Co L2,3-edge x-ray absorption spectra, (b) normalized oxygen K-edge x-ray absorption spectra of Li1−xCoO2 with respect to the cut-off voltage in the TEY mode, and ((c) and (d)) magnified parts of Co L3 and O K pre-edge spectra above 4.4 V, respectively, which are convenient for comparison. |
The evolution of surficial O is revealed by the TEY spectra of O K-edge in Fig.
Like the case in the bulk, the O evolution above 4.4 V draws more attention. Through the careful comparison between 4.4 V and 4.6 V shown in Fig.
3.4. Correlation between capacity fading and evolution of electronic structure
To shed light on the mechanism of capacity fading from the viewpoint of electronic structure, the sXAS spectra of LiCoO2 cathodes with different capacity retention ratios are studied, where the samples cycled between 3 V and 4.6 V are taken for example because of the most severe capacity degradation. Since the O K-edge is more sensitive to the evolution than the Co L-edge, only the TEY and TFY spectra of the O K-edge are shown.
The TFY data of the LiCoO2 cathodes in the 1st cycle are shown in Fig.
 | Fig. 4. (color online) (a) TFY and (b) TEY spectra of O K-edge of LiCoO2 cathodes in the 1st cycle, (c) TFY and (d) TEY spectra of O K-edge of LiCoO2 cathodes at fully charged state or fully discharged state with various capacity retention ratios. |
The capacity degradation during cycling is further studied. As shown in Fig.
The TEY data are shown in Fig.
The spectra of O K-edge of LiCoO2 in fully charged state cycled 10 times with cutoff voltages of 4.4 V and 4.6 V are compared. As shown in Fig.
 | Fig. 5. (color online) (a) TFY and (b) TEY spectra of O K-edge of LiCoO2 cathodes after 10 cycles with cutoff voltages of 4.4 V and 4.6 V. |
4. Conclusions
In summary, the charge compensation and capacity degradation are studied by the soft x-ray absorption spectroscopy at Co L2,3 and O K-edge in TFY and TEY modes.
The charge compensation from the oxidation of Co3+ is below 4.4 V while only O provide electrons above 4.4 V. The local O 2p holes are observed and its evolution gives an explanation to the origin of 
The residual Co4+ in the bulk is detected with LiCoO2 discharged to 3 V in the 1st cycle. More Co4+ ions appear on the surface as well as a bit of lithium salts are generated, which indicates that the interface undergoes more severe evolution including side reactions. By comparing the samples with various capacity retention ratios, Co–O bonds at the fully charged states are always found to be similar regardless of capacity degradation. In fully discharged states, a large number of Co–O bonds seem to be pinned at the fully charged states, where the mechanism needs further study. The similarity between the fully discharged states with ratios of 72% and 50% indicates that the structural change in the bulk is less related to the capacity degradation. The gradual aggregation of lithium salts on the surface, which is related to the decomposition and re-formation of the surface layer, is found to be more important for the capacity degradation. The present study helps to understand the aggravating capacity degradation with cutoff voltage increasing from 4.4 V to 4.6 V.
Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] | |
| [33] | |
| [34] | |
| [35] | |
| [36] | |
| [37] | |
| [38] | |
| [39] | |
| [40] | |
| [41] | |
| [42] | |
| [43] | |
| [44] | |
| [45] | |
| [46] | |
| [47] | |
| [48] | |
| [49] | |
| [50] | |
| [51] | |
| [52] | |
| [53] | |
| [54] | |
| [55] | |
| [56] |