



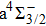


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Laser cooling of CH molecule: Insights from ab initio study
Cite this Article
Cui Jie, Xu Jian-Gang, Qi Jian-Xia, Dou Ge, Zhang Yun-Guang. Laser cooling of CH molecule: Insights from ab initio study. Chinese Physics B, 2018, 27(10): 103101 
Permissions
Laser cooling of CH molecule: Insights from ab initio study
† Corresponding author. E-mail:
Project supported by the National Natural Science Foundation of China (Grant No. 61705182).
Abstract
The feasibility of laser cooling a CH molecule is investigated theoretically by employing the ab initio method. The potential energy curves for the five Λ–S states and eight Ω states of CH are determined by the multi-reference configuration interaction with the Davidson corrections (MRCI+Q) level of theory. The results agree well with the available experimental data and other theoretical values. Also, the permanent dipole moments and transition dipole moments of the CH molecule are calculated at the multi-reference configuration interaction (MRCI) level. We find highly diagonally distributed Franck–Condon factors (f00 = 0.9950 and 0.9998) and branching ratios (R00 = 0.983 and 0.993) for the A2Δ → X2Π and C2Σ+ → X2Π transitions. Moreover, the values of suitable radiative lifetime τ of the A2Δ and C2Σ+ states are evaluated to be 9.64 × 10−7 s and 2.02 × 10−7 s, respectively, for rapid laser cooling. A scheme for laser cooling the CH molecule is designed. In the proposed cooling scheme, three wavelengths for A2Δ → X2Π and C2Σ+ → X2Π transitions are used, and the main pump lasers are λ00 = 430.86 nm and 313.45 nm, respectively. The feasibility of laser cooling the CH molecules is demonstrated for each of these schemes, and this study offers a theoretical basis for experimental research into preparation of cold CH molecules.
1. Introduction
The production of ultracold polar molecules can be useful in several emerging fields such as quantum simulation,[1,2] chemical dynamics,[3] and new platforms for quantum computing,[4] as well as in controlling chemistry.[5] Consequently, the interest in potential molecular candidates for laser cooling has grown in response to the commencement of investigation and experimental research in this field. As early as 2004, Di Rosa[6] conducted a brief survey of candidate molecules for laser cooling and identified a series of hydrides and halides, including CaH, AlH, AlF, AlCl, etc. However, it was thought to be difficult to directly laser cool molecules because of their complex structures until the direct laser cooling of SrF molecules was first achieved and reported by Shuman et al. in 2010.[7] Since then, successful laser cooling experiments have been performed for YO, CaF, BH, and BaH.[8–12] Based on existing investigations, it can be concluded that molecular candidates for laser cooling must possess highly-diagonal Franck–Condon factors (FCFs) and short radiative lifetimes. In practice, highly-diagonal FCFs mean that a limited number of lasers is required to keep the molecule in a closed-loop cooling cycle, and short lifetimes can ensure high spontaneous emission.
Very recently, laser cooling of diatomic polar molecules and molecular ions has begun to attract the attention of researchers. In published research, it was predicted that some alkaline-earth-metal hydrides and halides, such as BeH, MgH, BeCl, could be laser-cooled.[13–17] These molecules have one thing in common: they have no intervening state between the proposed transition for the laser cooling. Therefore, it is necessary to ensure that if an intervening state exists, there is nonetheless closed cycling between the upper state and the ground state of the transition. The A1Π → X1Σ+ and a3Π → X1Σ+ transitions of AlF were studied by Wells and Lane,[18] these two transitions both have highly diagonal FCFs, and the lifetimes are also extremely short. Fortunately, the emission rate of a3Π state is very small. Soon after this study, laser cooling of the SiO+ molecular ion was investigated by Nguyen and Odom[19] who found that between B2Σ+ and X2Σ+ states, there exits an intervening state, A2Σ+, but, due to its short lifetime, this did not prohibit the possibility of laser cooling. Following the publication of this study, increasing numbers of reports[20] of similar molecules, in which there exist intervening states, have provided bases for further research.
Here in this research, we report the accurate calculation of spectroscopic parameters and transition properties for the CH molecule to ensure its suitability for laser cooling. The CH radical occurring ubiquitously in interstellar space,[21] and high-power laser photochemistry[22] have been the subjects of numerous theoretical and experimental investigations. The experimental values of spin–orbit splitting for the X2Σ and A2Δ states of the CH molecule are 27.95 cm−1 and 1.0 cm−1, respectively, which were derived by Huber and Herzberg,[23] in 1979. In recent years, the spectroscopic constants of the X2Σ and A2Δ states were calculated by using different theoretical methods.[24–27] In 2003, Sun and Freed[28] used an ab initio effective valence shell Hamiltonian method to carry out a theoretical investigation of the spin–orbit splitting, and their results for the energies of these two states are 33.96 cm−1 and 1.27 cm−1, respectively. In these previous theoretical and experimental studies,[24–27,29–34] for the A2Δ → X2Σ transition, the region of transition energy (Te) from the A2Δ state to the X2Σ state was between 23147.884 cm−1 and 23565.4 cm−1; for the C2Σ+ → X2Σ transition, the region of Te was from 31802.128 cm−1 to 32205.44 cm−1. However, to the best of our knowledge, a systematic study of the laser cooling of CH is unavailable so far. The objective of the present work is to design a viable scheme to facilitate the laser cooling of the CH molecule, including computation of permanent dipole moments (PDMs), transition dipole moments (TDMs), FCFs, radiative lifetimes, and wavelengths for the A2Δ → X2Σ and C2Σ+ → X2Σ transitions of CH molecule by using a high-level ab initio method.
The rest of this paper is arranged as follows. In Section
2. Computational details
By using the MOLPRO 2015 program package,[35] all electronic structures of CH molecules for the X2Σ, a4Σ−, A2Δ, B2Σ−, and C2Σ+ states are calculated. In the first step we used the Post restricted Hartree–Fock (RHF) method to calculate the energy for the ground state. The next step was to obtain a multi-reference wavefunction by using the complete active space self-consistent-field (CASSCF) method.[36–38] The final step was based on the zero-order function, by using the multi-reference configuration interaction (MRCI) plus Davidson corrections (MRCI+Q) to calculate energies for the CH molecule,[39–42] and taking advantage of the Douglas–Kroll (DHK) method for correction.[43–45] The spin–orbit coupling (SOC) effects were also taken into consideration in the MRCI+Q calculations of the CH molecule.
Due to the limitation of the summary of the MOLPRO program package, the computations were performed within the C2v point group symmetry, which has four irreducible representations (irreps) (A1, B1, B2, and A2). The A1 irrep yields the Σ+ state and a component of Δ states, B1 provides the Π state, and A2 yields the Σ− state and the other components of Δ states. In the CASSCF calculations, six molecule orbitals are chosen as the active space, including four A1, one B1, and one B2. Five electrons are distributed in the (4110) active space. The aug-cc-pV5Z basis set is chosen for the hydrogen atom, and the aug-cc-pCV5Z basis set is for the carbon atom. In all electronic structure calculations, the aug-cc-pCV5Z (ACV5Z) basis set is used for the Λ–S and Ω states.
For the bound Λ–S and Ω states, all potential energy curves (PECs) are calculated with an interval of 0.05 nm over the distance of 0.04 nm–1.5 nm. To achieve the accurate results, the interval value is reduced to 0.02 nm, nearly the equilibrium bond distance. The spectroscopic constants are calculated by solving the nuclear Schrödinger equation through using the LEVEL 8.2 program, including the equilibrium bond length, Re, harmonic and an harmonic vibrational constants, ωe and ωe χe, respectively, rotational constant, Be, and adiabatic relative electronic energy referred to the ground state, Te.[46] With the PECs and TDMs of the different electronic states, this program can also yield the FCFs and Einstein coefficients for all possible discrete transitions allowed by selection rules. The Einstein coefficient Av′v for the rate of spontaneous emission from an initial rovibrational level (v, J) into the final rovibrational level (v′, J′) is defined by the expression[47]


3. Results and discussion
3.1. PECs and spectroscopic constants of Λ–S states
To determine whether the CH molecule is suitable for laser cooling, we must calculate the PECs and spectroscopic constants. At the MRCI+Q level, the CH molecule for the lowest two dissociation channels, C(3P) + H(2S) and C(1D) + H(2S), and the X2Σ, a4Σ−, A2Δ, B2Σ−, and C2Σ+ states are obtained. Figure
 | Fig. 1. (color online) PECs for the five Λ–S states of CH molecule. |
| Table 1. Spectroscopic constants for Λ–S states of CH molecule at MRCI+Q level. . |
As for the ground state X2Σ, the equilibrium bond distance and the vibrational frequency are calculated to be Re = 1.1181 Å and ωe = 2859.4759 cm−1, and their corresponding differences from experimental values are only 0.0019 Å and 1.3241 cm−1, respectively:[29,30] the percentage errors are only 0.17% and 0.05%, respectively. Compared with other theoretical values[24–27] of ωe χe and Be, our constants ωe χe = 62.3206 cm−1 and Be = 14.5084 cm−1 are in good accordance with the experimental data of 64.4 cm−1 and 14.457 cm−1. Our computed value for De is 3.669 eV, while the experimental value is 3.472 eV, and the percentage error is 5.67%. For the second singlet excited state B2Σ−, in the case of the spectroscopic constants Re, and Te, our data, 1.1624 Å, 26007.57 cm−1, respectively, are in better agreement with the experimental data (1.164 Å, 26059.517 cm−1) than those of other published calculations.[32,33] The value of ωe that we computed for the B2Σ− state is larger than the experimental measurement, giving a percentage error of 20.67%, but is in good agreement with other theoretical values, and the corresponding percentage error, between our theoretical value and others in the literature, for the B2Σ− state is 0.05% (Ref. [24]) ≤ δ ωe/ωe ≤ 1.85% (Ref. [25]).
For the first singlet excited state A2Δ of the higher dissociation channel C(1D) + H(2S), shown in Fig.
In conclusion, our spectroscopic parameter results agree with those of the experimental data and theoretical results in terms of the X2Σ, a4Σ−, A2Δ, B2Σ−, and C2Σ+ states of CH.
3.2. PECs and spectroscopic constants of Ω states
For the CH molecule, the spin–orbit coupling (SOC) effect makes the five Λ–S states split into eight Ω states, i.e. four Ω = 1/2 states, three Ω = 3/2 states, and one Ω = 5/2 state. Four new dissociation limits,C(3P0) + H(2S1/2),C(3P1) + H(2S1/2), C(3P2) + H(2S1/2), and C(1D2) + H(2S1/2) are generated from the original C(3P) + H(2S) and C(1D) + H(2S) limits. The PECs for X2Σ1/2, X2Σ3/2, 

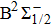
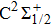
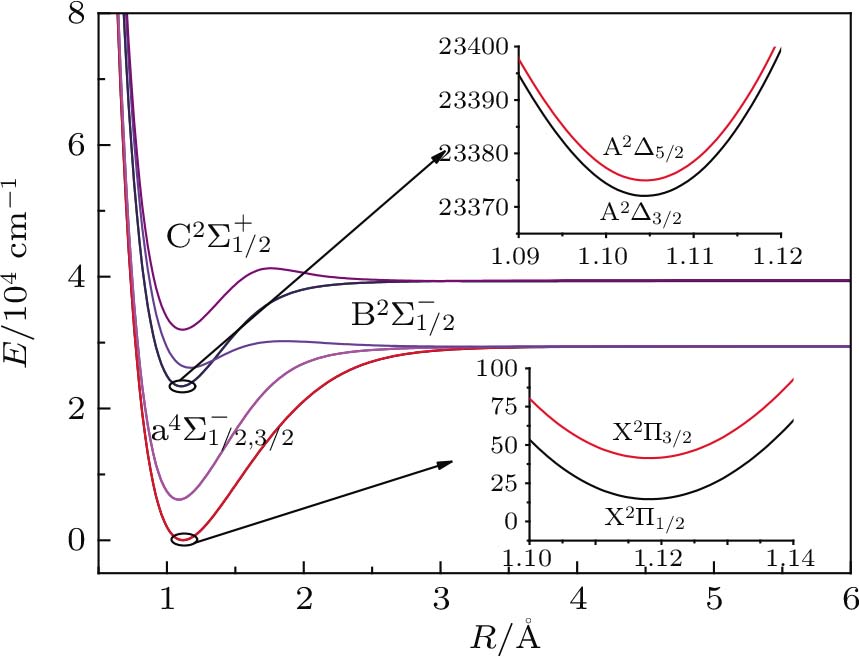 | Fig. 2. (color online) PECs for eight Ω states for the CH molecule. |
| Table 2. Spectroscopic constants of various bound Ω states of CH molecule. . |
As we can see from Table 

Owing to the SOC effect, the Λ–S X2Σ state splits into two X2Σ1/2 and X2Σ3/2 states. For the two Ω states, the energy sequence from low to high is 1/2, 3/2, and the small difference of Te between the states is 26.81 cm−1. By comparing the X2Σ state and the X2Σ1/2, X2Σ3/2 states in Table 

3.3. PDMs and TDMs
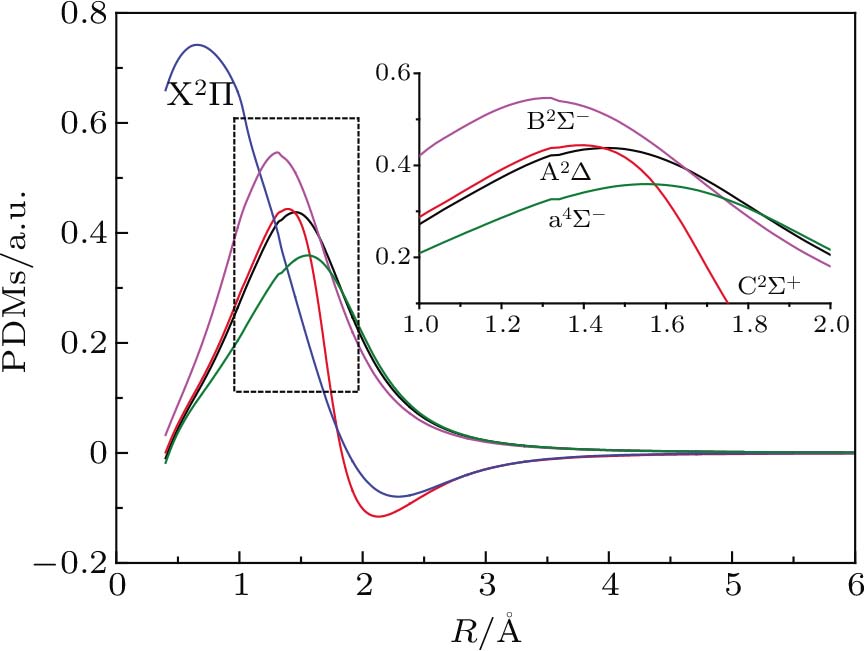
PDMs for the X2Σ, a4Σ−, A2Δ, B2Σ−, and C2Σ+ states are plotted in Fig.
 | Fig. 3. (color online) PDMs for five Λ–S states of CH molecule. |
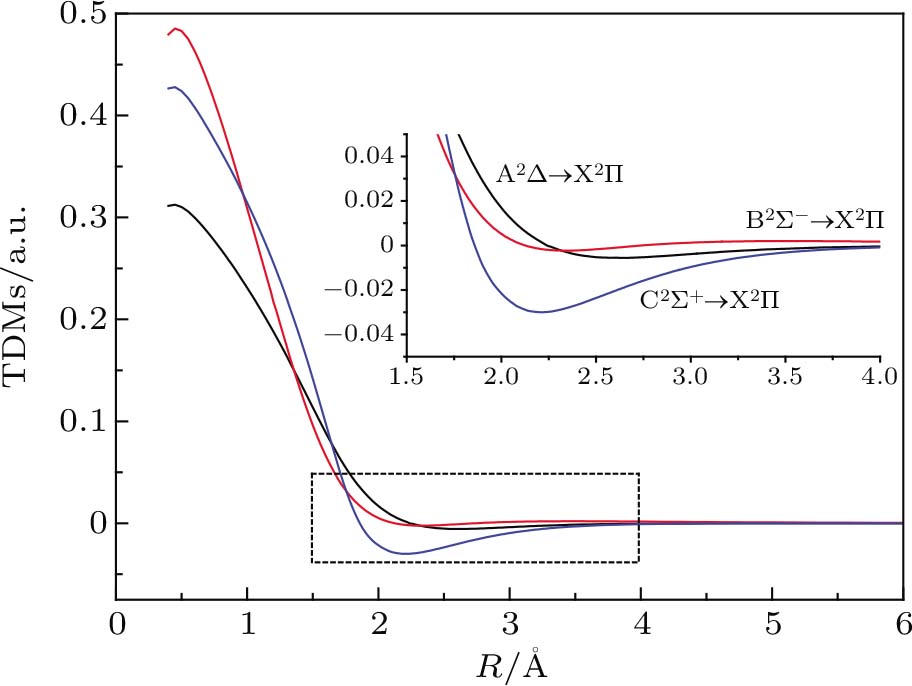
The TDMs for the A2Δ → X2Σ, B2Σ− → X2Σ, and C2Σ+ → X2Σ transitions as a function of the internuclear distance at the MRCI+Q level for the CH molecule are plotted in Fig.
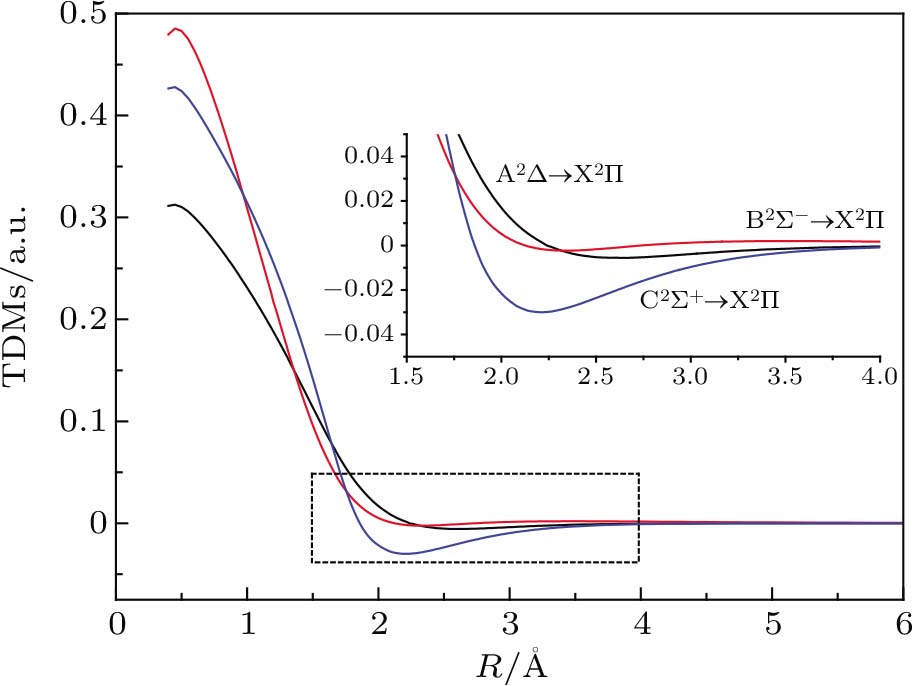 | Fig. 4. (color online) TDMs for A2Δ → X2Σ, B2Σ−→ X2Σ, and C2Σ+ → X2Σ transitions of CH molecule. |
3.4. Laser cooling of CH
As mentioned in Section
| Table 3. Calculated values of FCF fv′v, wavelength λv′v, and spontaneous radiative lifetime τ. . |
Comparing the relevant FCFs is a simple method of determining the cooling cycle, but it is not sufficient alone. The reason is that the relation between the relative strengths of the vibrational branching ratios and that between photon loss pathways are more direct than FCFs in the cooling cycle.[50–52] Thus, the cooling cycle with branching ratio Rv′v, which can be calculated by using the ratio of Einstein coefficients Av′v for each vibronic transition, is expressed as Rv′v = Av′v/ΣvAv′v. The calculated values of Einstein coefficient Av′v and vibrational branching ratio Rv′v of the A2Δ → X2Σ and C2Σ+ → X2Σ transitions are listed in Table
| Table 4. Calculated values of Einstein coefficient Av′v and vibrational branching ratio Rv′v of A2Δ → X2Σ and C2Σ+ → X2Π transitions. . |
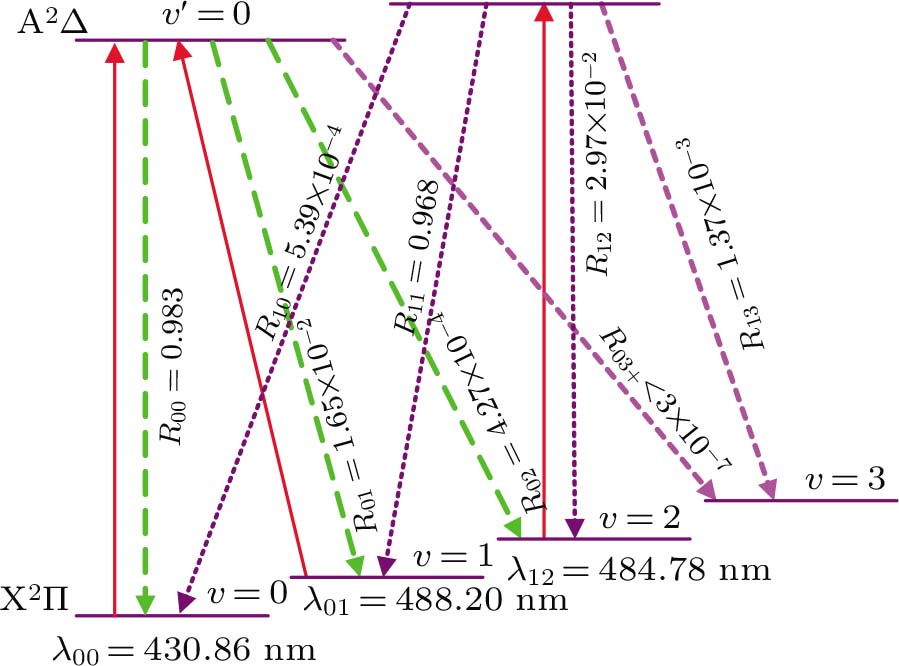
In Figs.
 | Fig. 5. (color online) Proposed laser cooling scheme for CH using A2Δ (v′)→ X2Π(v) transition (solid red) and spontaneous decay (dotted line) with calculated values of Rv′v. Here λv′v is the wavelength of A2Δ (v′) → X2Π(v) transition. |
 | Fig. 6. (color online) Proposed laser cooling scheme for CH using the C2Σ+ (v′) → X2Π(v) transition (solid red) and spontaneous decay (dotted line) with calculated values of Rv′v. Here Av′v is the wavelength of the C2Σ+ (v′) → X2Π(v) transition. |
4. Conclusions
The PECs for the Λ–S states X2Σ, a4Σ−, A2Δ, B2Σ−, and C2Σ+, and the Ω states X2Σ1/2, X2Σ3/2, 
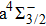

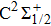
In Subsection
Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] | |
| [33] | |
| [34] | |
| [35] | |
| [36] | |
| [37] | |
| [38] | |
| [39] | |
| [40] | |
| [41] | |
| [42] | |
| [43] | |
| [44] | |
| [45] | |
| [46] | |
| [47] | |
| [48] | |
| [49] | |
| [50] | |
| [51] | |
| [52] |