





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Adsorptions of metal adatoms on graphene-like BC3 and their rich electronic properties: A first-principles study
Cite this Article
Sui Pengfei, Dai Jiaqi, Zhao Yinchang, Dai Zhenhong. Adsorptions of metal adatoms on graphene-like BC3 and their rich electronic properties: A first-principles study. Chinese Physics B, 2018, 27(9): 097311 
Permissions
Adsorptions of metal adatoms on graphene-like BC3 and their rich electronic properties: A first-principles study
† Corresponding author. E-mail:
Project supported by the National Natural Science Foundation of China (Grant Nos. 11774396 and 11704322) and Shandong Natural Science Funds for Doctoral Program, China (Grant No. ZR2017BA017).
Abstract
Density functional calculations have been performed to investigate the adsorption of twenty two different kinds of metal adatoms on graphene-like BC3. In contrast to the graphene adsorbed with adatoms, the BC3 with adatoms shows many interesting properties. (1) The interaction between the metal adatoms and the BC3 sheet is remarkably strong. The Li, Na, K, and Ca possess the binding energies larger than the cohesive energies of their corresponding bulk metals. (2) The Li, Na, and K adatoms form approximately ideal ionic bonds with BC3, while the Be, Mg, and Ca adatoms form ionic bonds with BC3 with slight hybridization of covalent bonds. The Al, Ga, In, Sn, and all transition metal adatoms form covalent bonds with BC3. (3) For all the structures studied, there exhibit metal, half-metal, semiconducting, and spin-semiconducting behaviors. Especially, the BC3 with Co adatom shows a quantum anomalous Hall (QAH) phase with a Chern number of −1 based on local density approximation calculations. (4) For Li, Na, K, Ca, Ga, In, Sn, Ti, V, Cr, Ni, Pd, and Pt, there exists a trend that the adatom species with lower ionization potential have lower work function. Our results indicate the potential applications of functionalization of BC3 with metal adatoms.
1. Introduction
Graphene, a two-dimensional honeycomb lattice of carbon, has stimulated a large number of research activities since its experimental realization.[1] So far, many exceptional features, such as the high strength of the lattice structure, massless Dirac fermions, the high thermal conductivity, and the half-integer Hall conductance, have been revealed for this atomically thin layer.[2–5] To modify the electronic properties of graphene, recent investigations have confirmed some chemical functionalized graphene structures like graphane,[6–10] grapheneoxide,[11–13] fluorographene,[14–18] etc. Meanwhile, the adsorption of metal atoms is also used to change electronic structures to enhance the applications of graphene. For instance, the adsorption of alkali and alkali-earth metal adatoms can dope electrons to graphene, resulting in a down-shift of the Dirac point;[19] metal-coated graphene possesses considerable hydrogen storage capacity;[20–22] lithium decorated graphene shows conventional superconductivity;[23] the graphene with 5d transition metal adatoms presents quantum anomalous Hall (QAH) effect,[24] etc. However, the binding energies (Eb) between the metal atoms and graphene are lower than the cohesive energies (Ec) of the corresponding bulk metals,[19,25,26] which implies that the clustering of the adatoms is inevitable. In order to increase Eb, it is proposed to substitute the carbon atoms in graphene with some metal atoms.[27] Nonetheless, the substitution of carbon atoms with metal atoms creates vacancies and damages the structure of graphene severely.[28]
It was reported that the boron doped carbon nanostructures could suppress the clustering of metal adatoms.[29–32] Most strikingly, at the same time of the production of graphene,[1] the BC3 honeycomb sheet was also realized experimentally.[33,34] Different from graphene, the hexagonal BC3 sheet is a semiconductor due to the lack of dangling bonds in it. For hydrogen storage, this two-dimensional (2D) BC3 sheet has been predicted as one of the most promising host materials because there is no clustering of metal adatoms.[35,36] Recently, the Fermi surface nesting in the BC3 was investigated, revealing that Li coated BC3 can be taken as an experimentally feasible structure for exploring the nesting-driven quantum phases.[37] Furthermore, the topological superconductivity in doped BC3 sheet was also predicted, which provides a promising route toward the realization of a genuine 2D helical p+ip superconductor.[38] Therefore, it is important to systematically investigate the adsorption of metal atoms on the BC3 sheet. In this paper, twenty two different metal adatoms, whose adsorptions on graphene or silicene have been studied,[19,39,40] are used to study their adsorption properties on the BC3 sheet. The binding energy, geometry, density of states (DOS), dipole moment, charge transfer, and work function of each adadatom–BC3 structure are calculated. The calculation details are described in Section
2. Method
The spin-polarized density functional theory (DFT) is employed in our calculations by using the Vienna ab initio simulation package (VASP) code.[41] The projector augmented-wave (PAW) method[42] is adopted with an energy cutoff of 520 eV. The generalized gradient approximation (GGA) with Perdew–Burke–Ernzerhof (PBE)[43] exchange correlation energy is used. For the pristine BC3 unit cell, in which there are two boron atoms and six carbon atoms, we use the Brillouin zone (BZ) samplings with 12 × 12 × 1, 18 × 18 × 1, and 36 × 36 × 1 Monkhorst–Pack grids[44] for relaxations, static calculations, and density of state calculations, respectively. The Monkhorst–Pack grid for a BC3 supercell is chosen to be inversely proportional to the dimensions of the supercell. The vacuum space along the z direction is larger than 15 Å. The tolerance for the energy convergence is 10−5 eV and all structures are fully relaxed until the force on each atom is smaller than 0.01 eV/Å. The dipole corrections implemented in VASP code[45,46] are applied to potentials and total energies in all systems to remove the spurious dipole interactions between the periodic images.
3. Results and discussion
The bond lengths of B–C and C–C bonds in the BC3 sheet are 1.56 Å and 1.42 Å, respectively, which is consistent with the previous work.[47,48] The lattice constant of BC3 unit cell is 5.17 Å which is larger than twice of that of graphene due to the existence of the B–C bonds. The electronic structure shown in Fig.
 | Fig. 1. (color online) The band structure, projected density of states (PDOS), and band decomposed charge densities of VB and CB at the Γ, M, and K symmetry points for pristine BC3 unit cell. The Fermi level is set to be at zero. |
A supercell consisting of (2 × 2) BC3 unit cells presented in Fig.
 | Fig. 2. (color online) Top view and lateral view of the (2 × 2) structure of the hexagonal BC3. The big and the small balls represent the boron and carbon atoms, respectively. Six different adsorption-sites are labeled as H1, H2, T1, T2, B1, and B2. |
3.1. Energetic, structural, and electronic properties
For each adatom, the adsorption on the six possible sites are investigated firstly, as shown in Fig.
 | Fig. 3. (color online) Binding energy of 22 different metal adatoms on six possible adsorption sites of the (2 × 2) BC3 sheet. The data on sites of H1, B1, T1, B2, T2, and H2 are listed from left to right for each adatom studied. |
3.1.1. Alkali and alkali-earth metal adatoms
For Li, Na, K, and Ca, the binding energy Eb on any adsorption site is larger than the cohesive energy Ec per atom of the corresponding bulk metal, as shown in Fig.
| Table 1. Properties of the metal atom adsorption on the BC3 sheet. FS represents the favorite sites of adatoms on the BC3, h is the adatom height defined as the difference between the adatom z coordinate and the average z coordinate of atoms in BC3, Δ is the adsorption-induced distortion deviating from the plane of BC3, ρad is the charge transfer from adatom to BC3, IP is the experimental ionization potential of the isolated atom, μiso and μtot are the magnetic moment of the isolated atom and the total magnetic moment of the system, p and Φ are the dipole moment and the work function, and Egap is the energy gap of the system. The “m” and “h” in Egap column represent metal and half-metal, respectively. . |
Figure
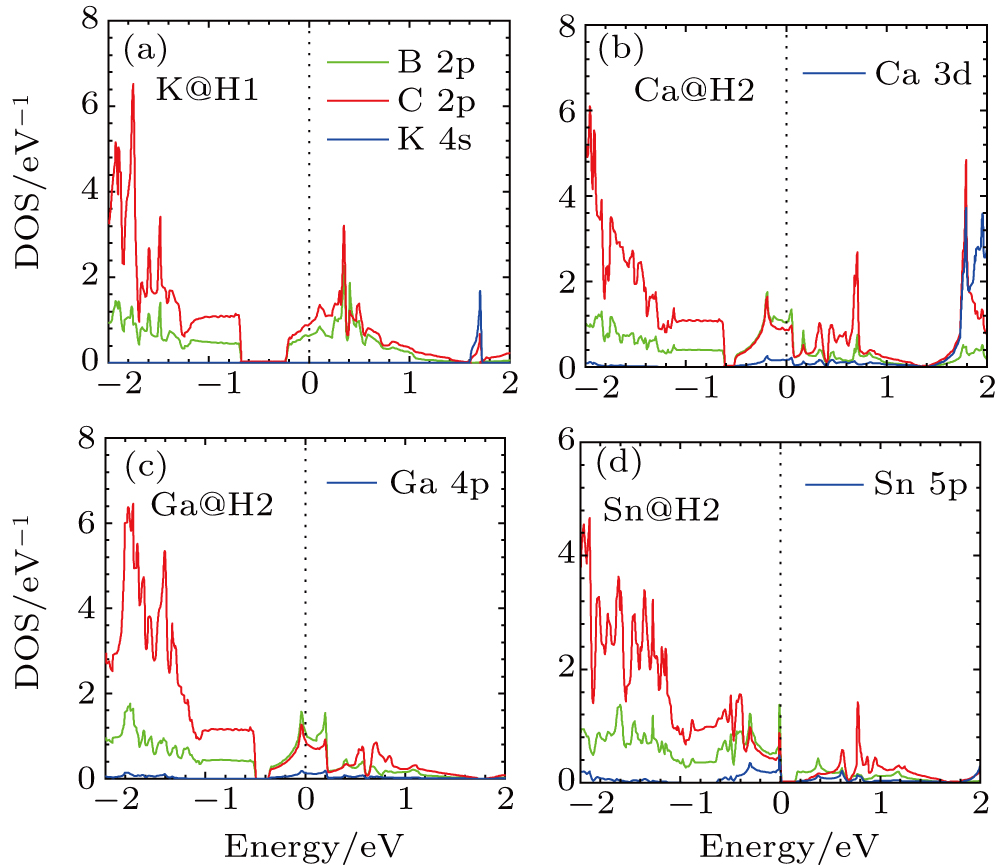 | Fig. 4. (color online) PDOS for (a) K–BC3, (b) Ca–BC3, (c) Ga–BC3, and (d) Sn–BC3. |
For the adsorption of Be, Mg, and Ca atoms on BC3, slight hybridization states occur close to EF as shown in Fig.
3.1.2. Groups III and IV metal adatoms
The Eb of Al, Ga, and In on the BC3 sheet are almost three times as large as on graphene. Furthermore, the Eb of Sn adatom on BC3 is 2.75 eV with the ratio Eb/Ec of 0.88, which is an order of magnitude larger than that on graphene.[19] Due to the similar atomic radii of Al, Ga, In, and Sn, the adatom heights h of the four adatoms vary from 2.00 Å to 2.35 Å, and their dipole moments vary from 0.71 debye to 2.01 debye. The charge transfer from Al, Ga, In, and Sn adatoms to the substrate are 0.71 e, 0.51 e, 0.58 e, and 0.71 e, respectively, which are much less than the corresponding values of the valence electrons. There are remarkable hybridizations between the adatom p states and the substrate states around and below EF for Al, Ga, and In on BC3. Figure
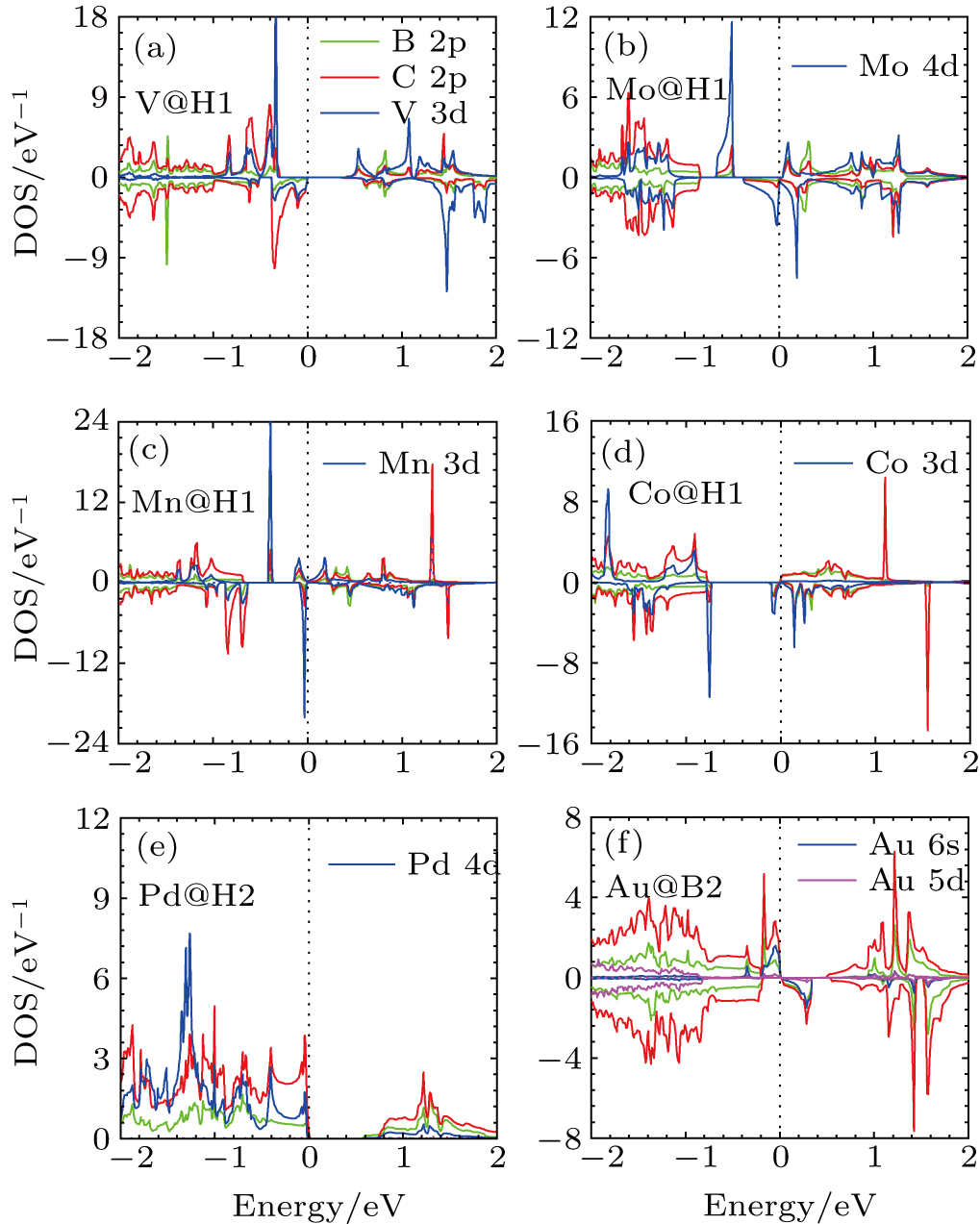
3.1.3. Transition and noble metal adatoms
For the adsorption of the transition and noble metal atoms on the BC3 sheet, the adatoms are strongly bounded to the H1 or H2 sites except for Au, which prefers to bond with the substrate at the B2 site. Among these transition metal adatoms, the Eb of Ti adatom is close to Ec, such that the ratio Eb/Ec is 0.93. The adatom d states are strongly hybridized with the B 2p and C 2p states of BC3 around EF for all these adatoms except Au, whose 6s states are hybridized with the substrate states near EF, as shown in Fig.
 | Fig. 5. (color online) PDOS for (a) V–BC3, (b) Mo–BC3, (c) Mn–BC3, (d) Co–BC3, (e) Pd–BC3, and (f) Au–BC3. |
In details, figure
More interesting are the cases of Co, Mo, Mn, and Au adatoms on BC3. In Co–BC3 structure, there exists a single spin Dirac cone around EF, as shown in Figs.
 | Fig. 6. (color online) Band structures for Co at the H1 site of the BC3 calculated with the (a) GGA, (b) LDA, and (c) LDA with SOC. (d), (e), and (f) show band structures for Mo–BC3, Mn–BC3, and Au–BC3, respectively. |
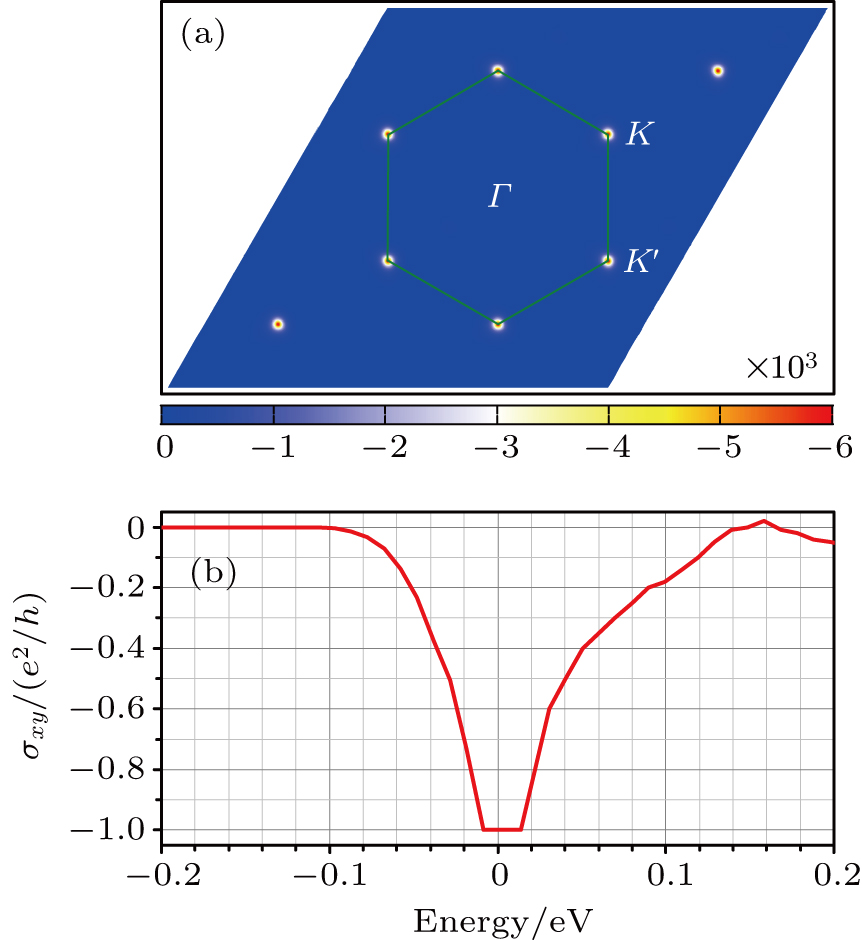
For the Co–BC3 system, the ground state estimated from the LDA calculations is found to be ferromagnetic, which is about 211 meV and 45 meV lower than the nonmagnetic and antiferromagnetic states, respectively. To identify the topological property of the SOC induced insulating state, we also calculate the Berry curvature Ω(k) and Chern number for the Co–BC3 system.[57–59] The distribution of the Berry curvature Ω(k) (in atomic units a.u.) for the whole valence bands in the momentum space is shown in the top panel in Fig.
 | Fig. 7. (color online) (a) The distribution of the Berry curvature (in a.u.) for the whole valence bands in momentum space and (b) the anomalous Hall conductivity σxy around the Fermi level calculated from the LDA. |
3.2. Relation of dipole moments vs. work functions
The dipole moment perpendicular to the substrate is determined by the real-space-charge rearrangement due to the interaction between the adatom and substrate. It is defined as[19]p = −∫ρ(z)zdz+ΣiZiezi, where ρ(z) is the electron density integrated over the xy-plane, i is the index of ion, Zi is the atomic number of ion, and zi is the z coordinate of ion. The work function, which denotes the energy to remove an electron from the adatom–substrate system, can be calculated as Φ = Evac−EF, where Evac is the reference vacuum energy which is determined from the electrostatic potential in the vacuum region. Thus the work function is sensitive to the charge transfer from adsorbate to substrate.
To reveal the correlations between the dipole moment p and the work function Φ, we have drawn the p–Φ plot in Fig.
 | Fig. 8. (color online) (a) The dipole moments p vs. the work functions Φ and (b) the IP vs. the work function Φ for all adatoms adsorbing on their most stable sites. |
4. Conclusion
In summary, we have studied the adsorption properties of alkali, alkaline-earth, groups III–IV, and transition metals on the graphene-like BC3 sheet. Many interesting properties are concluded. (1) The interactions between the adatoms and the BC3 are quite strong in contrast to those found in adatoms-graphene. (2) For Li, Na, and K on BC3, the adatom s states lie far above from EF, which results in the ionic bond for these adatoms. For Be, Mg, and Ca adatoms, besides the ionic bonding, there exist hybridizations between adatoms and the substrate near EF. The Al, Ga, In, and Sn adatoms are covalently bounded with the nearest neighboring atoms of the BC3 sheet. The adatom d states of all transition metal atoms studied are strongly hybridized with the BC3 states around EF except for Au, whose s states are hybridized with the substrate near EF. (3) BC3 with Co adatom reveals a quantum anomalous Hall (QAH) insulator phase as inferred from LDA calculations. (4) For Mo and Mn adatoms on BC3, the structures exhibit half metal behavior, while the most stable Au–BC3 structure is spin-semiconductor. (5) For Li, Na, K, Ca, Ga, In, Sn, Ti, V, Cr, Ni, Pd, and Pt, there is a trend that adatom species with lower IP have lower Φ. These results indicate the potential applications of the functionalization of graphene-like BC3 with metal adatoms.
Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] | |
| [33] | |
| [34] | |
| [35] | |
| [36] | |
| [37] | |
| [38] | |
| [39] | |
| [40] | |
| [41] | |
| [42] | |
| [43] | |
| [44] | |
| [45] | |
| [46] | |
| [47] | |
| [48] | |
| [49] | |
| [50] | |
| [51] | |
| [52] | |
| [53] | |
| [54] | |
| [55] | |
| [56] | |
| [57] | |
| [58] | |
| [59] |