






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Density functional study on the bimetallic TimZrn (n + m ≤ 5) clusters and their interactions with H2*
Project supported by the Scientific Research Plan Foundation of Sichuan Education Department of China (Grant No. 2014JY0072).
|
|

Project supported by the Scientific Research Plan Foundation of Sichuan Education Department of China (Grant No. 2014JY0072).

Project supported by the Scientific Research Plan Foundation of Sichuan Education Department of China (Grant No. 2014JY0072).
† Corresponding author. E-mail:
Project supported by the Scientific Research Plan Foundation of Sichuan Education Department of China (Grant No. 2014JY0072).
Equilibrium geometries, stabilities, and electronic properties of small TimZrn (n + m ≤ 5) clusters were investigated using the density functional method. The ground states were determined, and it was found that the larger clusters and those consisting of more Zr atoms are more stable. The electronic properties of the clusters were discussed based on HOMO–LUMO gaps, vertical ionization potentials (VIP), and vertical electron affinities (VEA). Furthermore, we studied the interactions between those clusters and molecular hydrogen, and found that in all the cases dissociative chemisorptions occurred. According to the chemisorption energies, the pure Zr clusters are relatively more active towards H2 when compared with the others except Ti3Zr, which shows the highest activity. The magnetic moments of TimZrn and TimZrnH2 were also compared, and the results show that the hydrogenated clusters have the same or decreased total magnetic moments with respect to the bare clusters except for Ti3Zr2.
In the past few decades, a great deal of attention has been paid to metallic clusters due to their unique physical and chemical properties, which can be drastically different from those of their bulk counterparts.[1–4] Investigations on clusters can also build a bridge for people to understand the evolution from atoms to bulk systems. With vast application prospects, studying the atom aggregation process and cluster stability is of great significance, about which theoretical methods can provide more details compared with experimental approaches.[5–7] While studying pure atomic clusters leads to fundamental understanding of the quantum size effects on the properties of the clusters, bimetallic clusters consisting of two different atoms have been found to have properties that differ from those of the pure constituent atom clusters.[8–12] Furthermore, by changing the cluster size and the atom number, some properties of a cluster may be adjusted for different applications.
As transition metal (TM) elements, zirconium and titanium have been extensively explored as structural and functional materials. Alloys consisting of Ti and Zr have huge potential in many fields such as biomedicine,[13] dentistry,[14] catalysts,[15] and storage systems.[16,17] Titanium and zirconium are 3d and 4d TM elements, which belong to the same family, with the outer electron configurations of 3d24s2 and 4s24p64d2, respectively. Closeness and spatial overlap of d orbitals along with s and p orbitals usually make their interatomic reactions complicated.[18] Concerning their nanoclusters, many studies have been focused on them under the framework of the density functional theory (DFT). The evolutions of monoatomic Ti and Zr clusters have been extensively studied,[19–24] and their stable geometries, energetic, and electronic properties were the main topics. Those reports have reached the consensus that Ti7, Ti13, Ti15, and Zr7 are magic clusters. Moreover, it was found that many small clusters possess net spin magnetic moments, which is thought to be a common phenomenon for TM clusters. As a further step, some researchers have researched the doped and mixed Ti and Zr clusters using first-principle theories.[25–29] Besides, the investigations concerning the interactions of hydrogen molecules with the two kinds of bare clusters revealed that H2 prefers a dissociative chemisorption on them.[30–33] Many similar theoretical studies on the adsorption or chemisorption of H2 onto small clusters have been reported, which help people understand the catalytic mechanisms on surfaces and contribute to the development of clusters in hydrogen storage materials.[34–37]
However, studies of bimetallic Ti–Zr clusters have been rarely reported. To the best of our knowledge, only Wang et al.[38] carried out a fundamental investigation on (TiZr)n (n = 1–7) clusters, and obtained their stable structures, bonding properties, infrared spectra, and so on. No study on Ti–Zr cluster systems with varied chemical compositions is available. Therefore, in this paper, we present a systematic study of the TimZrn (n + m ≤ 5) clusters. Furthermore, the chemisorption behaviors of H2 on them are studied, which we hope can provide information for hydrogen storage applications.
This paper is organized as follows. In Section
The geometry optimizations and vibrational frequency analyses of bare and hydrogenated TimZrn (n + m ≤ 5) clusters were performed by using the DFT method as implemented in the Gaussian program series. First, a variety of possible initial geometries of every cluster were established in GuassView 3.07. Then, those geometries were optimized in Gaussian 03, and the structures would change to the stable ones. The stable structures were obtained in the absence of imaginary frequency by computing the vibrational frequencies at the same theory level. Besides, all the geometries were optimized at different possible spin states. The lowest-energy structure of a certain cluster was defined as the ground state cluster.
It is of great significance to choose a relatively accurate method to do computational calculations. In the present work, we adopted def2-TZVP,[39] a triple-zeta basis set, to treat all the atoms. To find an appropriate functional, we optimized Ti2, Zr2, and TiH dimers with a variety of exchange–correlation functionals, and compared the results with the values derived from experiments[40–43] as listed in Table
| Table 1. Calculated and experimental equilibrium distances (R), vibrational frequencies (ω), and dissociation energies (ED) of the diatomic clusters. . |
We first optimized the geometries of the bare TimZrn (n + m ≤ 5) clusters. The most stable structures together with their spin multiplicities and point groups are shown in Fig.
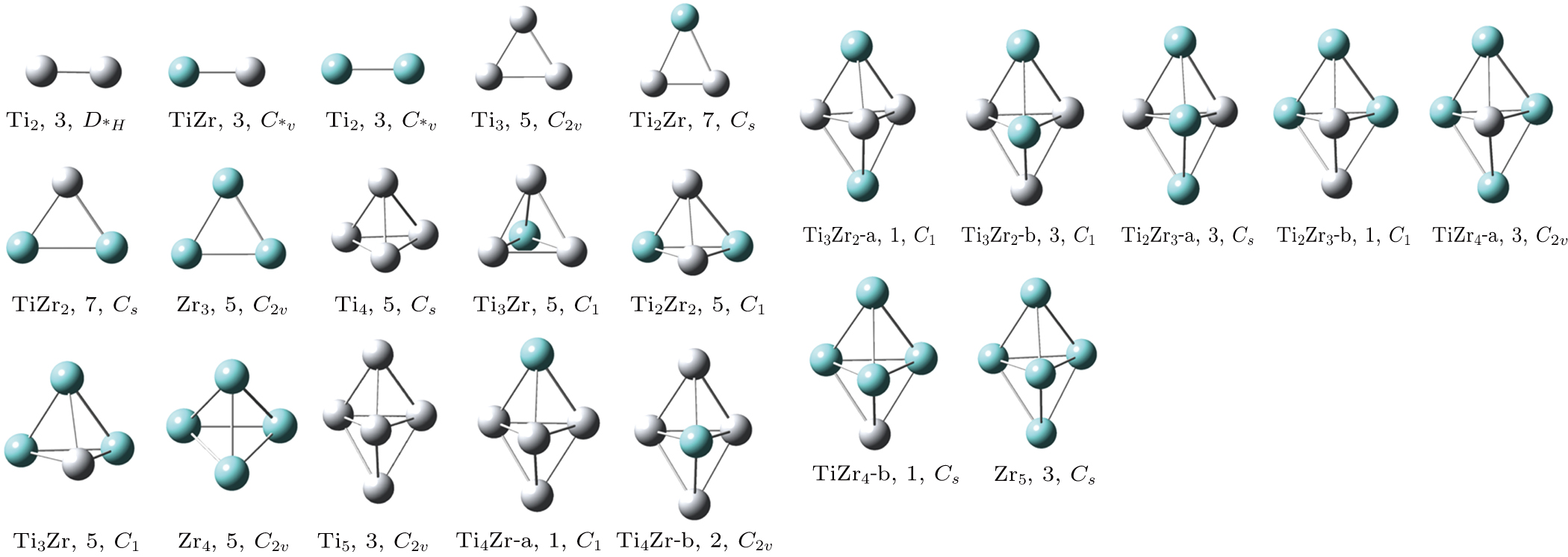 | Fig. 1. (color online) Equilibrium structures, spin multiplicities, and point group symmetries of the TimZrn (n + m ≤ 5) clusters. The gray balls and blue balls represent Ti and Zr atoms, respectively. |
From the above, the TimZrn (n + m ≤ 5) clusters can be seen as deriving from the Zr atoms taking the places of the original Ti atoms in the titanium clusters or vice versa. This may imply that the two metal elements are compatible and substitutable in some cases.
Now we discuss the structural stability, which can usually be described by the binding energy per atom (Eb). The Eb is calculated according to
 |
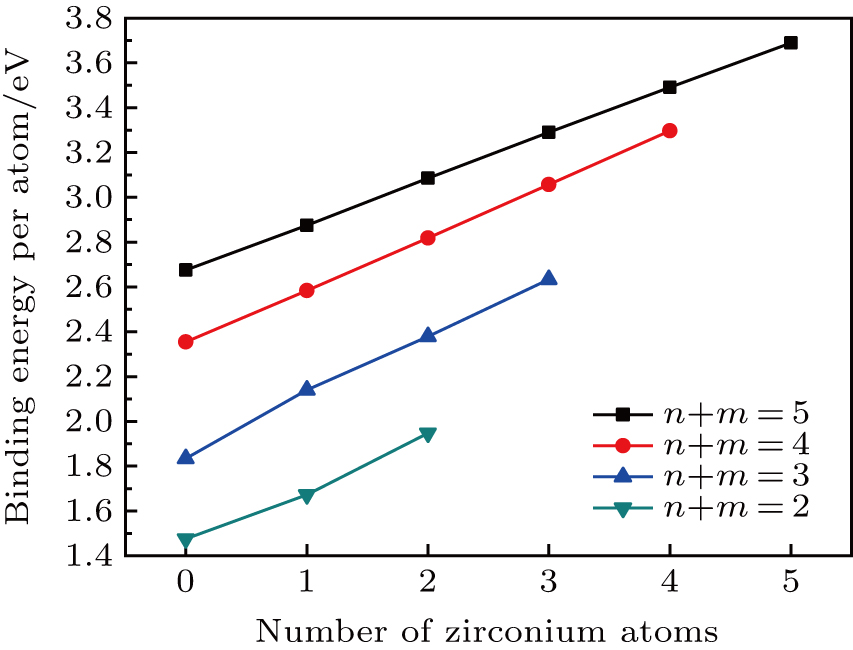 | Fig. 2. (color online) Variations of binding energies as a function of the number of Zr atoms for the TimZrn (n + m ≤ 5) clusters. |
| Table 2. Binding energies (Eb), HOMO, LUMO, HOMO–LUMO gaps (Eg), VIP, and VEA of the TimZrn (n + m ≤ 5) clusters in units of eV. . |
The dissociation energy is another indicator to describe the stability of clusters, which is defined as follows:
 |
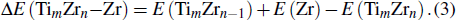 |
 | Fig. 3. (color online) Variations of dissociation energies towards a Ti atom as a function of the number of Ti atoms for the TimZrn (n + m ≤ 5) clusters. |
 | Fig. 4. (color online) Variations of dissociation energies towards a Zr atom as a function of the number of Zr atoms for the TimZrn (n + m ≤ 5) clusters. |
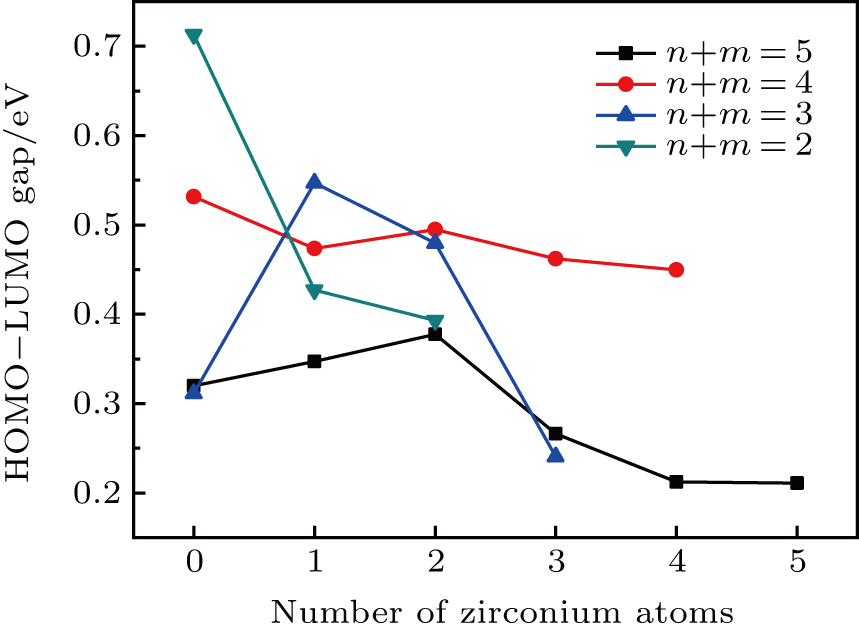
The energy difference between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), i.e., the HOMO–LUMO gap, reflects the ability of an electron to be excited from an occupied orbital to an unoccupied orbital. Generally speaking, a larger HOMO–LUMO gap is related to enhanced chemical stability. The variation of the HOMO–LUMO gaps of the clusters versus the number of zirconium atoms is plotted in Fig.
 | Fig. 5. (color online) HOMO–LUMO energy gaps as a function of the number of Zr atoms for the TimZrn (n + m ≤ 5) clusters. |
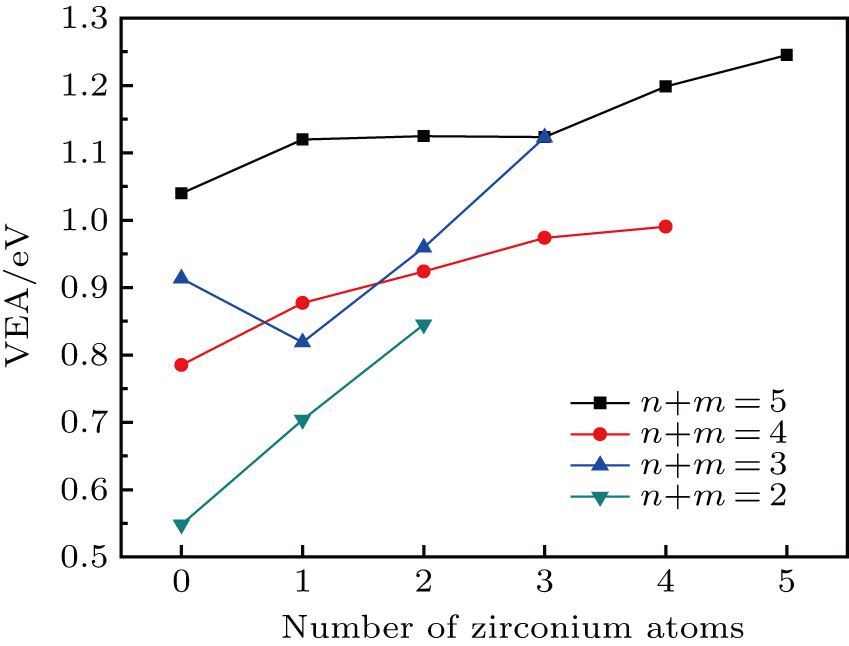
The ionization potential and electron affinity are another two significant parameters for the evaluation of chemical stability. In the present work, we have obtained the vertical ionization potentials (VIP) and the vertical electron affinities (VEA) of the TimZrn clusters, which are respectively calculated by
 |
 |
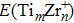

 | Fig. 6. (color online) Vertical ionization potentials as a function of the number of Zr atoms for the TimZrn (n + m ≤ 5) clusters. |
 | Fig. 7. (color online) Vertical electron affinities as a function of the number of Zr atoms for the TimZrn (n + m ≤ 5) clusters. |
One of the main motives of our current work is to investigate the chemical activity of the TimZrn (n + m ≤ 5) clusters towards molecular hydrogen. We performed an exhaustive search in order to find the minimum-energy structures for the hydrogenated clusters. A hydrogen molecule was introduced to each cluster from various directions and distances, and the stable TimZrn structures in both the lowest and the second lowest energies were used in this step. The lowest-energy structures after hydrogenation as well as their spin states and point groups are displayed in Fig.
 | Fig. 8. (color online) Equilibrium structures, spin multiplicities, and point group symmetries of the hydrogenated TimZrn (n + m ≤ 5) clusters. The big gray balls, big blue balls, and small gray balls represent Ti, Zr, and H atoms, respectively. |
| Table 3. Chemisorption energies towards H2 (ΔECE), H–H distances (RH−H), and charges of the Ti–Zr host clusters and H atoms for the TimZrnH2 (n + m ≤ 5) clusters. . |
The activity of H2 on these clusters can be investigated by calculating the chemisorption energies for the systems according to
 |
 | Fig. 9. (color online) Variations of H2 chemisorption energies as a function of the number of Zr atoms for the TimZrnH2 (n + m ≤ 5) systems. |
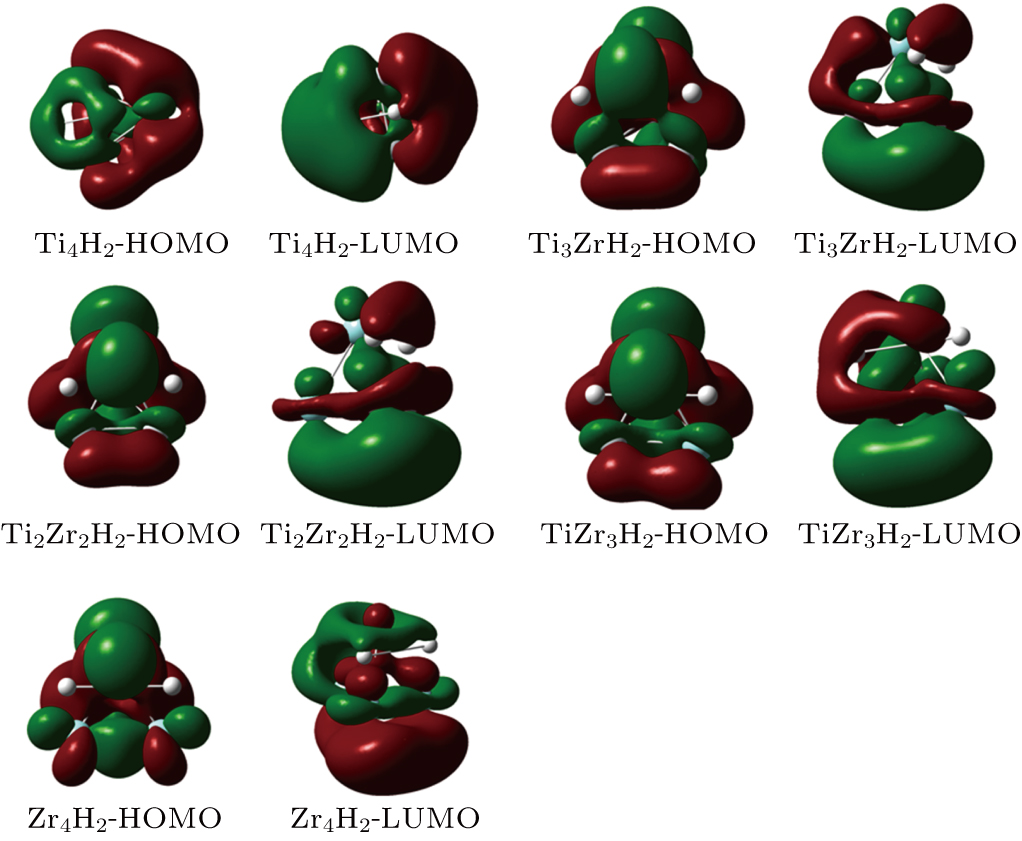
In Fig.
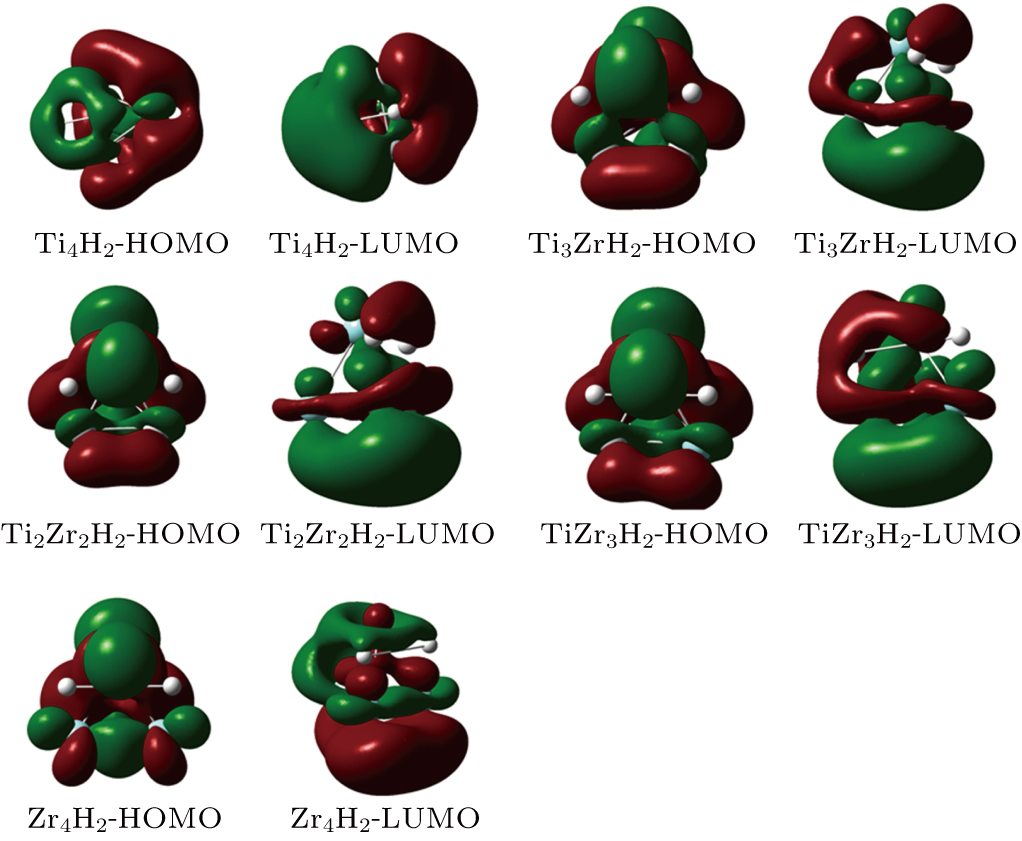 | Fig. 10. (color online) HOMO and LUMO distributions of the TimZrnH2 (n + m = 4) clusters. |
We finally discuss the magnetic properties of the TimZrnH2 clusters. To compare the hydrogenated clusters with the bare clusters, their spin magnetic moments are listed together in Table
| Table 4. Magnetic moments of the bare (μ0) and hydrogenated (μ) TimZrn (n + m ≤ 5) clusters. . |
The equilibrium geometries, stabilities, and electronic properties of small TimZrn (n + m ≤ 5) clusters were investigated by using density functional calculations combining the PBE functional with the def2-TZVP basis set. The ground state structures of the clusters of the same size are similar to each other. According to the averaged binding energies, the clusters become more stable as the cluster size gets larger and as the number of Zr atoms increases. The clusters consisting of more Ti atoms are usually characterized by larger HOMO–LUMO gaps and VIP as well as smaller VEA, implying their electronic structures are of higher stability. Moreover, the interactions between molecular hydrogen and the stable TimZrn clusters were investigated. Our results show that H2 prefers dissociative chemisorptions onto the bridge or face sites of the considered clusters. We calculated the chemisorption energies of these systems and found the pure Zr clusters are more active towards H2 when compared with the Ti counterparts. The maximum of ΔECE appears at the Ti3ZrH2 cluster, which may be understood by analyzing their HOMO and LUMO states, when strong interactions among the H atoms and the host cluster can be observed. Besides, the total spin magnetic moments of these clusters have decreased or remained the same after hydrogenation except for Ti3Zr2.
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] | |
| [33] | |
| [34] | |
| [35] | |
| [36] | |
| [37] | |
| [38] | |
| [39] | |
| [40] | |
| [41] | |
| [42] | |
| [43] |