


{kind=link}
{kind=link}
{kind=link}
Diffusion Monte Carlo calculations on LaB molecule
Cite this Article
Elkahwagy Nagat, Ismail Atif, Maize S M A, Mahmoud K R. Diffusion Monte Carlo calculations on LaB molecule. Chinese Physics B, 2018, 27(9): 093102 
Permissions
Diffusion Monte Carlo calculations on LaB molecule
† Corresponding author. E-mail:
Abstract
Potential energy curves for the lowest electronic states of LaB and LaB− have been calculated by ab initio calculations. The diffusion Monte Carlo method has been employed in combination with three different trial functions. Spectroscopic constants have also been numerically derived for the neutral molecule and compared with the only available theoretical work;[19] however, predictions are provided for the corresponding constants for the anionic species which have not been reported yet. Our calculations suggest the high spin quintet state of LaB as the ground state with the triplet state higher in energy irrespective of the type of the functional used. This suggestion is in good accordance with the previous theoretical results calculated at B3LYP/LANL2DZ level of theory, whereas it contradicts with the prediction based upon B3LYP/SDD calculations in the same study. Moreover, variations of the permanent dipole moments as a function of the internuclear separations for the two electronic states of the neutral molecule have been studied and analyzed.
1. Introduction
Over the years, systems containing rare earths have received much attention in many fields of physics and chemistry. As far as the ab initio calculations are concerned, several problems have probably obstructed accurate theoretical treatments for these systems. On one hand, relativistic and electron correlation effects become important in heavy elements and should not be neglected in accurate calculations. On the other hand, there are several open shells with different main and angular quantum numbers, i.e. 4f, 5d, and 6 s, whose orbitals are comparable in energies and therefore the bonding can take place with any of these orbitals, which adds additional complexity for quantum chemical calculations for systems involving these elements.
Understanding the electronic structures of lanthanide diatomics is of great importance for studying lanthanide compounds. Lanthanum diatomics, in particular, are good candidates for providing useful information on more complex systems involving lanthanides, owing to their simpler open d shell electronic configurations. Several research groups have performed ab initio calculations and experimental investigations on diatomic lanthanum molecules. Lanthanum monohydride, for instance, has been extensively studied in the past, both theoretically[1–8] and experimentally.[9–12] Spectroscopes of lanthanum halides[1,13–17] and lanthanum monoxide[1,3,4,18] have also been thoroughly investigated during the past few decades. However, studying diatomic lanthanum boride, which is the focus of this study, is scarce in literature. To our knowledge, there is no experimental study up to now on lanthanum boride. Only recently, a theoretical study has been reported on the 5d-transition metals monoborides by Kalamse et al.[19] In their paper, the authors have done the first theoretical study on lanthanum monoboride using density functional method based B3LYP functional with LANL2DZ and SDD basis sets. They have also calculated the lowest spin states, bond lengths, electron affinities, ionization potentials, and binding energies of diatomic lanthanum boride. However, no potential curves for LaB states have been reported. Unfortunately, Kalamse and co-workers have predicted in their work two different ground states for LaB based on the basis set used. They have predicted that LaB has triplet ground state at B3LYP/SDD level whereas it is quintet using B3LYP/LANL2DZ method.
The confusion provided by the previous study for assigning the LaB ground state as well as the lack of reliable information on lanthanide boride are the motivations of our present work, in which we apply the diffusion Monte Carlo (DMC) method to study the neutral and anionic LaB. Nowadays, DMC is considered as a promising method for treating the strongly correlated electron systems. Besides its favorable scaling with system size, it explicitly includes electron-electron correlation effects. A primary goal of this study is to provide the first calculations of potential energy curves (PECs) for neutral and anionic LaB molecules and to determine the nature of the ground states of them as well. The effects of different trial wavefunctions are also examined using single determinants constructed from HF orbital as well as from density functional theory (DFT) orbitals with the commonly used B3LYP and B3PW91 functionals. This work is also relevant to study the dipole moment dependence on internuclear distances for the two states of the neutral LaB molecule. An estimation of the electron affinity of LaB is also the subject of this work.
2. Computational methods
where the parameter β defines the asymmetry of the Morse potential, R is the internuclear distance. Re and De are the equilibrium bond length and the dissociation energy, respectively, which were numerically derived from the fitting process. Finally, accurate calculations on the ground state energies of neutral LaB and anion at the equilibrium bond lengths were carried out in order to obtain computational estimate of the electron affinity by difference.
All computations for energies and dipole moments presented in this paper were carried out via the diffusion Monte Carlo method by making use of the QWalk package.[20] The diffusion Monte Carlo method has been extensively described in the literatures, so we only refer the reader interested in details to Refs. [21]–[23]. In our DMC calculations, the trial wavefunction was generated using the quantum chemistry program Gamess.[24] We propose to employ the DFT-type trial wavefunctions instead of using highly expensive correlated functions. Two density functionals were employed: (i) the Becke three parameters hybrid exchange (B3) + LYP correlation (B3LYP),[25–27] (ii) the Becke three parameter hybrid exchange (B3) + PW91 correlation (B3PW91).[25,28] In addition, HF trial wavefunction was also included for the sake of comparison. The basis set used in our calculations for lanthanum atom is CRENBS ECP,[29] where 54 core electrons are replaced by effective potentials. However, for the boron atom we utilized the pseudopotential of Burkatzki et al.[30] produced for use with quantum Monte Carlo. Through all the DMC calculations a time step of τ = 0.001 H−1 and a mean population of 2000 walkers were used which proved to be successful for the calculations.
To construct the potential energy curves, we perform an accurate fit of the discrete DMC computed values of the total energy via the Morse potential function with the analytical form
 |
3. Results and discussion
3.1. Potential energy curves and spectroscopic constants
The calculated DMC potential energy curves of LaB in triplet and quintet states using the three different functionals are presented in Fig.
 | Fig. 1. DMC potential energy curves of the triplet and quintet states for neutral LaB molecule using different functionals. (a) HF, (b) B3LYP, and (c) B3PW91. The unit a.u. is short for atomic units. |
The derived bond lengths, dissociation energies, and transition energies for the two electronic states are summarized in Table
| Table 1. Bond lengths Re, dissociation energies De, and transition energies Te computed within DMC for the triplet and quintet states of LaB molecule together with the available reference data. . |
On the other hand, one can see that our estimated DMC bond lengths are in good accordance with that reported previously using LANL2DZ basis set. However, our estimated value of the same quantity for the triplet state is significantly shorter than the previous result calculated via SDD basis set. We believe that the multireference character of the triplet state is the main source of error in the previous B3LYP calculations for such state. In fact, the multireference triplet state is poorly dominated by single reference methods. This interpretation is confirmed in the study of Č ernušák and co-workers[42] on scandium boride. In their work, the quintet state was well described by a single reference method. However, the triplet state gave unacceptable results using the same strategy, confirming the multireference character for the latter state. The different basis sets utilized in the previous B3LYP calculations might be also partially responsible for the contradictory prediction of LaB ground state. On the other hand, the bond length and the corresponding dissociation energy are most likely underestimated at DMC/HF, which is an expected consequence of using HF trial function. As we stated before, HF orbitals have been utilized here for comparative purposes only.
It may be seen from Table
Now we turn the attention to the anionic LaB which is treated with the same ansatz like its neutral counterpart. The PECs are displayed in Fig.
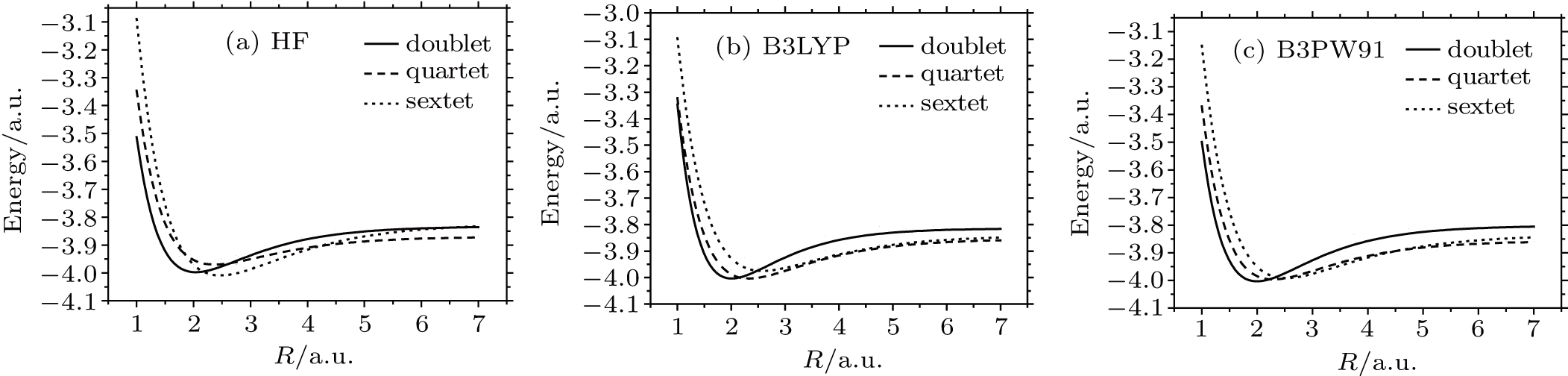 | Fig. 2. DMC potential energy curves of the doublet, quartet, and sextet states for LaB− molecule using different functionals. (a) HF, (b) B3LYP, and (c) B3PW91. |
| Table 2. Bond lengths Re, dissociation energies De, and transition energies Te computed within DMC for the doublet, quartet, and sextet states of LaB−. . |
Now it seems appropriate to compute the electron affinity (EA) of LaB molecule. Table
| Table 3. Electron affinity of LaB molecule computed with DMC method using different functionals together with the available reference data. . |
3.2. Dipole moments
It is well known that the dipole moment is a sensitive tool to test the accuracy of both the calculated wavefunction and energy. Theoretical predictions of the dipole moments of lanthanide diatomics, in particular, depend strongly on the level of treatment of the electron correlation. Our goal in this section is to investigate the permanent dipole moment for the lowest two states of LaB molecule as a function of internuclear distances using both DFT functionals as well as HF. This is presented graphically in Fig.
 | Fig. 3. The permanent dipole moments of (a) the triplet and (b) the quintet states of LaB molecule computed with the DMC method using different functionals. |
It is still interesting to compare the dipole moment values obtained at equilibrium separations for the two states of LaB along with the data in Ref. [19]. It is apparent from Table
| Table 4. The permanent dipole moment μ of the triplet and quintet states for LaB molecule computed with DMC method using different functionals together with the available reference data. . |
4. Conclusion
In the present work, we have computed the potential energy curves for the lowest electronic states of LaB and LaB− by means of the DMC method employing three different functionals. The PECs are accurately fitted to Morse potential and the spectroscopic constants are numerically derived. Our calculations conclude that the ground state of LaB is most likely to be the high spin quintet state in line with the previous theoretical results calculated at B3LYP/LANL2DZ level of theory, whereas it contradicts with the prediction based upon B3LYP/SDD calculations in the same study. Our estimated quintet states bond lengths, employing the three functionals, compare well to the value obtained by the previous study; meanwhile, our calculations yield triplet bond lengths which are considerably longer. In addition, our results of De for the two states provide a significant overestimation compared with the earlier values.
On the other hand, the ground state for LaB− has been determined and the corresponding spectroscopic constants have been reported for the first time. This work further studies the dipole moment distribution for the two lying states of the neutral molecule as a function of the internuclear separations. Our results concerning the neutral LaB and anion are entirely new, and will certainly be helpful for guiding future experimental and theoretical spectroscopic works.
Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] | |
| [33] | |
| [34] | |
| [35] | |
| [36] | |
| [37] | |
| [38] | |
| [39] | |
| [40] | |
| [41] | |
| [42] | |
| [43] | |
| [44] | |
| [45] | |
| [46] | |
| [47] | |
| [48] |