








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A simulation study of water property changes using geometrical alteration in SPC/E
Cite this Article
Li Ming-Ru, Zhang Nan, Zhang Feng-Shou. A simulation study of water property changes using geometrical alteration in SPC/E. Chinese Physics B, 2018, 27(8): 083103 
Permissions
A simulation study of water property changes using geometrical alteration in SPC/E
† Corresponding author. E-mail:
Project supported by the National Natural Science Foundation of China (Grant Nos. 11635003, 11025524, and 11161130520), the National Basic Research Program of China (Grant No. 2010CB832903), and the European Commission’s 7th Framework Programme (Fp7-PEOPLE-2010-IRSES) (Grant Agreement Project No. 269131).
Abstract
We present a systematic investigation of the impact of changing the geometry structure of the SPC/E water model by performing a series of molecular dynamic simulations at 1 bar (1 bar = 105 Pa) and 298.15 K. The geometric modification includes altering the H–O–H angle range from 90° to 115° and modifying the O–H length range from 0.90 Å to 1.10 Å in the SPC/E model. The former is achieved by keeping the dipole moment constant by modifying the O–H length, while in the latter only the O–H length is changed. With the larger bond length and angle, we find that the liquid shows a strong quadrupole interaction and high tetrahedral structure order parameter, resulting in the enhancement of the network structure of the liquid. When the bond length or angle is reduced, the hydrogen bond lifetime and self-diffusion constant decrease due to the weakening of the intermolecular interaction. We find that modifying the water molecular bond length leading to the variation of the intermolecular interaction strength is more intensive than changing the bond angle. Through calculating the average reduced density gradient and thermal fluctuation index, it is found that the scope of vdW interaction with neighbouring water molecules is inversely proportional to the change of the bond length and angle. The effect is mainly due to a significant change of the hydrogen bond network. To study the effect of water models as a solvent whose geometry has been modified, the solutions of ions in different solvent environments are examined by introducing NaCl. During the dissolving process, NaCl ions are ideally dissolved in SPC/E water and bond with natural water more easily than with other solvent models.
Keyword:molecular dynamics simulation;geometric modification;intermolecular interaction;ion’s solvation
1. Introduction
Water is the most important liquid for our existence. Despite the tiny size of the water molecule, a network structure can be made by strong intermolecular hydrogen bonds (H-bonds) in the liquid ambient condition.[1–4] This capacity to form H-bonds, except as the non-directional interactions seen in some simple liquids, results in many unusual properties. For instance, density exhibits as maximum at 4°C and viscosity shows decreases under high pressure.[5–8] These property changes are strongly related to the structure of the water liquid phase, which in turn depend on the intermolecular potential. Additionally, in aqueous solutions, water as a solvent also has a significant effect on the conformation of proteins and nucleic acids molecule, such as pleated sheets and helices.[9–12] Despite significant research into properties of water, its structural evolution is still somewhat unclear.[13–15] The problem is, how does a water molecule evolve to an optimal geometric structure? How does the intermolecular interaction change and is the solubility of water changed as the water molecule evolves to the optimal structure? Therefore, to explore the structural evolution of the water molecule is very important and intriguing. With the development of computer technology, molecular dynamics (MD) simulations become an important tool: they are not limited by the constraints of experiments, thus offering an ideal route for studying the individual contributions of different aspects.
Two types of modified water models used in such investigations include the alteration of either the geometric angle (Bent model) or the relative strength of electronic and dispersive forces alteration (Hybrid model). Lynden-Bell and colleagues performed a series of molecular dynamic simulations, which resulted in various interpretations regarding the properties of these modified liquids.[16–21] They did so by changing the SPC/E model’s H–O–H angles from 90° to 60° and adjusting the Lennard-Jones (LJ) parameters σ to produce approximately the same dipole moment parameters as the SPC/E water model. This resulted in the water’s network structure changing from a three-dimensional to a one-dimensional chain. The H-bonding strength was affected by altering the weight of the Lennard-Jones term relative to the electrostatic term. This leads to a change of the liquid structure from water-like to a new type of Lennard-Jones liquid without an evident local order structure.
Yet another study[22] focused on investigating the effects of altering the SPC/E model geometric angle as a function of temperature within the angle range from 120° to 60°. They found that the density anomalies appear as the bond angle is at least 100°. Increasing the bond angle leads to a shift of the density maximum anomaly. In addition, their study also showed that the first-neighbour shell for 60° is relatively changed over 200, 250, and 300 K; however, both 90° and 100° gradually acquire water-like structural features upon cooling.
It is inevitable that the change of a water molecule’s structure, and the hydrogen and oxygen atom charge lead to changing its properties. Nevertheless, the dissolving of charged ions strongly depends on the structure or property of the solvent around them. In our pervious research,[23] the relative weight of Lennard-Jones and electronic components was modified, based on the SPC water model, to investigate the relationship between the solution of NaCl and the H-bonding ability of water at room temperature and standard atmospheric pressure. The results indicated that NaCl is most ideally dissolved in normal water with the strongest hydration effects around both cations and anions.
The current study seeks to probe the property changes of water when the geometry structure of the water molecule is altered, and also tries to understand changes in the intermolecular interaction between water molecules during the geometric evolution of a water molecule. So we build a series of water models (not real water) based on the SPC/E water model. Similarly, the solvation behavior of the Na+ and Cl− ions in these water models is also investigated. The O–H lengths variation ranges from 0.90 Å to 1.10 Å (1 Å in SPC/E), while the change in H–O–H angles ranges from 90° to 115° (109.47° in SPC/E). The optimal distance between atoms is found to vary with the change in the SPC/E model geometric structure. The distance change, however, has a significant effect on the strength of H-bonding due to the variation of the inter-molecular interaction. The average reduced density gradient (aRDG) and thermal fluctuation index (TFI) methods are used further to study the change of intermolecular interaction in a dynamic environment and provide a good way to distinguish H-bond, vdW interaction, and steric effect. Finally, we consider the solvation of NaCl in these models. These studies can help us to further gain fundamental understanding of the microscopic origin of the water molecule during geometric evolution.
The remainder of this paper is organized as follows. In Section
2. Model and methods
2.1. Model
where Elj describes the Lennard-Jones interaction between two molecules, and Ec is the electrostatic interaction between two charged sites.
The SPC/E water model[24] is used for the simulation in conjunction with classical molecular dynamics simulations. The force field parameters of the SPC/E model, Na+ and Cl− ion are presented in Table
| Table 1. Values of Lennard-Jones and electrostatic interaction potential parameters. e represents the magnitude of the electronic charge. . |
| Table 2. Modified water models. ∠HOH, rOH, and rHH are respectively the water molecule’s bond angle, bond length, and distance between two hydrogen atoms. Model (I) represents the change of the H–O–H angle. Model (II) represents the change of the O–H length. . |
The potential energy E between two molecules is written as a sum of pairwise intermolecular interactions and can be expressed as
 |
2.2. Simulation details
In our simulations, the following molecular dynamic simulation protocol is adopted. Every simulation cell contains 256 water molecules for pure solvent in a cubic box. The v-rescale thermostat[26] and Berendsen barostat[27] are used to regulate the temperature T at 298.15 K and pressure P at 1 atm (1 atm = 1.01325 × 105 Pa), with time constants of 0.2 ps and 0.5 ps, respectively. The leapfrog algorithm with a time step size 1 fs and the SHAKE algorithm[28] are used respectively to integrate the equation of motion and constrain the intramolecular geometry for the rigid models. The cutoff radius of the Lennard-Jones and Coulombic interactions is 9 Å. The Particle Mesh Ewald (PME) method is used to treat electrostatic interactions.[29,30] In the ion solution, the molarity of NaCl is 2.3 M. Every simulation cell contains 234 solvent molecules, 11 Na+, and 11 Cl− ions with cubic periodic boundary conditions. To simulate the variations introduced by ions, we use the same ensemble conditions as pure solvents. Finally, the resulting 70-ns NPT simulation is carried out for each case using GROMACS software.[31]
2.3. Reduced density gradient (RDG) and thermal fluctuation index (TFI)
where ρ(r ) is the electron density and |∇ρ(r )| is the electron density gradient mode. The RDG is a dimensionless quantity describing the difference between the actual electron density and a homogeneous electron distribution. For the average reduced density gradient (aRDG), the average electron density

and average electron density gradient mode

can be obtained through calculating the dynamics trajectory.
where std [ρ(r )] is the standard deviation of electron density in a dynamical trajectory, which can be written as:
where n is the number of frames in consideration and ρi(r ) is the density calculated based on the geometry of frame i. All the different models iso-surfaces are plotted with the VMD code.[33]
The reduced density gradient (RDG) and the thermal fluctuation index (TFI) are carried out by the Multiwfn program.[32] The RDG can be written as follows:
 |
Furthermore, the stability of weak interaction between molecules can be given by the TFI based on the aRDG and is defined as:
 |
 |
2.4. Hydrogen bond lifetimes
with si(t) = {0, 1} for hydrogen bond i at time t. The integral of C(τ) gives an estimate of the average H-bonding lifetime τHB:
The H-bond lifetimes can be computed in different ways.[34,35] In the present study, we use a geometrical criterion to determine the existence of H-bonding as shown in Fig.
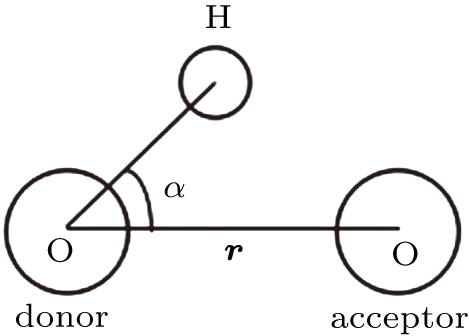 | Fig. 1. Geometrical hydrogen bond criterion. |
The value of 3.5 Å corresponds to the first minimum of the radial distribution functions (RDFs) of the SPC/E water model. The lifetime of the H-bonds is calculated from the average over all autocorrelation functions of the existence functions (either 0 or 1) of all H-bonds:
 |
 |
3. Results and discussion
3.1. Structure
The local structure of liquid can be conveniently studied by the radial distribution functions (RDFs). In Fig.
 | Fig. 2. The RDFs of (a) oxygen–oxygen and (b) oxygen–hydrogen atom pairs for various bond length models. |
 | Fig. 3. The RDFs of (a) oxygen–oxygen and (b) oxygen–hydrogen atom pairs for various bond angle models. |
 | Fig. 4. (color online) Spatial distribution functions of local oxygen density around the water molecule in the first shell in the various water models. It shows an isovalue of 10.5 for B115, compared to an isovalue of 5.5 for other water models. |
As shown in Fig.
Svishchev and Kusalik[36] used firstly the SDFs to study the local structure of water. Figure
As seen in Fig.
3.2. Local tetrahedral order and quadrupole interaction
where ψjk is the angle between the bond vectors r ij and r ik, with j and k representing the four nearest tetrahedral atoms, such that perfect tetrahedra corresponds to q = 1. In addition, these model’s quadrupole interactions QT can be measured using[41,42]
where 2θ is the H–O–H angle and μ is the dipole moment.
The parameters of orientational order ⟨q⟩ have often been used in recent years to investigate structural order in liquids and glasses.[37–40] The 
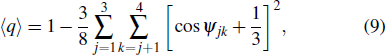 |
 |
As shown in Fig.
 | Fig. 5. Variation of the local order parameters ⟨q⟩ and quadrupolar interactions QT for various H–O–H angles, and O–H length models. |
The bond angle or length decrease, due to the weakening of the intermolecular attraction, resulting in the water molecule becoming more mobile. This indicates that the local tetrahedral order of water molecules is disrupted. Thus, ⟨q⟩ is reduced monotonically. In addition to the variation in gOO(r) [shown in Fig.
3.3. Self-diffusion constants and hydrogen-bond lifetime
with the correlation times ranging from 0 to 1 ns.
The self-diffusion constants of various models were calculated from their mean squared displacements
 |
The correlation of self-diffusion constants and H-bonding lifetime for various H–O–H angles and O–H length models are depicted in Fig.
 | Fig. 6. Variation of the self-diffusion constants and hydrogen bond lifetime for various H–O–H angles and O–H length models. |
To explore further the sensitivity to changes in bond length (elastic model) or bond angle (bent model), the data of the H-bonding lifetime in Fig.
3.4. Mean squared displacement
From the mean squared displacement (MSD) as a function of time, Fig.
 | Fig. 7. Mean squared displacement (MSD) as a function of time for various H–O–H angles and O–H length models. |
3.5. Local weak interaction
Weak interaction analysis between molecules or atoms, called non-covalent interaction analysis, has been recently developed by Contreras-Garcia et al.[44–47] It is based on an analysis of the electron density distribution in molecular systems in the regions of low electron density and low gradient. This approach is often referred to as reduced density gradient (RDG) analysis[37] and provides a good way to distinguish H-bond, vdW interaction, and steric effect.
Figure
 | Fig. 8. (color online) Average reduced density gradient (RDG) and thermal fluctuation index (TFI) figures of different O–H length and H–O–H angle water models, where panels (a) and (c) correspond to aRDG, (b) and (d) correspond to TFI, respectively. Both aRDG and TFI isovalue are 0.25, using Multiwfn software default parameters. For aRDG, the color depth, in blue, represents the electrostatic interaction or H-bonding strength in the corresponding region. Similarly, the deeper red color represents the more intensive steric effect. Green indicates a low electron density region, corresponding to vdW interaction. For TFI, more blue (red) means TFI is smaller (larger), and thus the weak interaction in the corresponding region is more stable (unstable). |
Furthermore, the green isosurface around a water molecule exhibits in which direction this water tends to interact with other waters by vdW interaction, as shown in Figs.
3.6. Ion–ion and ion–water correlations
In order to investigate the solution properties of water models with the geometry modified, ion solutions in different solvent environments are examined by adding NaCl ions. Use of the ion solution generally introduces two effects due to the ions on interactions: (i) it breaks the original solvent structures, making the solvent a little more mobile, and (ii) new hydration shells around ions reduce water’s diffusion constant.[49–52]
The RDFs of ion–ion and ion–water in the NaCl solutions of different solvents are given in Fig.
 | Fig. 9. RDFs between ion–ion and ion–water for NaCl solutions. Panels (a), (b), and (c) represent the H–O–H angle variation while panels (d), (e), and (f) represent the O–H length variation. |
4. Conclusion
In this work, we have studied the liquid structure, dynamic, water molecule local weak interaction, and NaCl’s solution behavior of modified water models at 1 bar and 298.15 K. These models, based on alteration of the SPC/E model, have O–H lengths ranging from 0.90 Å to 1.10 Å (the H–O–H angles do not change) and H–O–H angles ranging from 90° to 115° (the O–H length changes from 0.82 Å to 1.08 Å). Both types of models have the same atomic charge and LJ parameters as the original SPC/E model.
To summarize, we find that the properties of the liquids undergo dramatic change caused by altering the water molecule geometry structure. Increasing the H–O–H angle and O–H length leads to: the structure of liquid becoming more highly-structured, raising the order parameter and QT, longer H-bond lifetime, and shorter atoms distance adjoining water molecules. In contrast, decreasing the H–O–H angle and O–H length results in a gradual changing of the liquid structure into being more like a Lennard-Jones liquid with spherical symmetrical local arrangement. The liquid exhibits a glassy-like state feature, when the SPC/E’s O–H length is increased by 0.1 Å. All these variations can be primarily attributed to the alteration of the intermolecular interaction strength between water molecules. However, the change in the intermolecular interaction strength is very sensitive to changes in the bond length.
Using the weak interaction and thermal fluctuation analysis methods, some interesting regions are obtained around a central water molecule. The scope of vdW (van der Waals) interaction with neighboring water molecules is reduced as the bond length and angle are decreased, and vice versa. A water molecule as a donor forms H-bonds with other water molecules leading to more stability, whereas the stability slightly reduces when the water acts as an H-bond acceptor. In addition, for the NaCl dissolving process, SPC/E water is an ideal solvent which dissolves ions and more easily couples with Na+ and Cl− ions than in other solvent environment models.
This study helps gain further fundamental understanding of the property changes of a water molecule by altering the water molecule geometry structure. Ongoing research will investigate the conformation transitions of large molecules such as DNA, protein, etc., in these modified models.
Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] | |
| [33] | |
| [34] | |
| [35] | |
| [36] | |
| [37] | |
| [38] | |
| [39] | |
| [40] | |
| [41] | |
| [42] | |
| [43] | |
| [44] | |
| [45] | |
| [46] | |
| [47] | |
| [48] | |
| [49] | |
| [50] | |
| [51] | |
| [52] |