




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Delay time dependence of wave packet motion and population transfer of four-level K2 molecule in pump–pump–probe pulses
Cite this Article
Chang Zhiqiang, Li Changming, Guo Wei, Yao Hongbin. Delay time dependence of wave packet motion and population transfer of four-level K2 molecule in pump–pump–probe pulses. Chinese Physics B, 2018, 27(5): 053301 
Permissions
Delay time dependence of wave packet motion and population transfer of four-level K2 molecule in pump–pump–probe pulses
† Corresponding author. E-mail:
Abstract
The effect of delay time on photoelectron spectra and state populations of a four-level ladder K2 molecule is investigated by a pump1–pump2–probe pulse via the time-dependent wave packet approach. The periodical motion of the wave packet leads to the periodical change of the photoelectron spectra. The Autler–Townes triple splitting appears at zero delay time, double splitting appears at nonzero delay time between pump1 and pump2 pulses, and no splitting appears at nonzero delay time between pump2 and probe pulses. The periodical change of the state populations with the delay time may be due to the coupling effect between the two pulses. It is found that the selectivity of the state populations may be attained by regulating the delay time. The results can provide an important basis for realizing the optical control of molecules experimentally.
Keyword:wave packet motion;population transfer;intense femtosecond pump1-pump2-probe pulse;four-level ladder K2 molecule
1. Introduction
The femtosecond laser can regulate the molecular dynamics in real time. A benefit of light manipulation of molecular processes is controlling the evolution of the wave packet. The photoelectron spectra and state populations map the wave packet dynamics information of the excited state. They were studied in multi-level molecular systems, and were found to be sensitive to the parameters of the laser fields.[1–33]
For an alkali metal dimer, a femtosecond pump–probe pulse can be used for studying the dynamics of the wave packet on the excited state. Braun et al.,[1] Miao et al.,[2,3] and Liu et al.[4] found that the state populations and photoelectron spectra of NaI change with the delay time. Zhao et al.[5] studied the sensitivity of the predissociation dynamics of NaI to the field-free orientation. Yu et al.[6] studied the effect of the delay time and laser wavelength on the evolution of the wave packet of NaK. The periodical motion of the wave packet leads to the periodical change of the photoelectron spectra. Maet al.[7] found that the wave packet on the excited state moves periodically, the state population of NaRb is affected by the laser intensity. Zhang et al.[8,9] suggested that the laser wavelength affects the oscillation period of the wave packet, and the peak positions and heights of the photoelectron spectra of NaLi vary with the delay time. The efficiency of population transfer through diabatic coupling strength changes with the laser intensity and laser wavelength. The delay time alters the peak positions and heights of the photoelectron spectra. Zhu et al.[10] presented that the delay time affects the state populations of the CsI molecule. Meng et al.[11,12] and Wang et al.[13] studied the sensitivity of the state populations of NO to the laser intensity, laser wavelength, pulse width, and delay time, and presented the periodical change with the laser intensity and pulse width. Cheng et al.[14] found that the selective excitation of excited states can be achieved by phase modulation. Niu et al.[15] proposed that the rovibrational population transfer can be controlled by a two-overlapping-pulse scheme. Xie et al.[16] found that the population exchange can occur due to the driving action of an external field.
The dynamics of the wave packet of a homonuclear diatomic molecule shares some similar characteristics with that of a heteronuclear diatomic molecule. The periodical wave packet motion of the four-level Na2 was studied in Refs. [17]–[19]. Yuan et al.[20,21] found that the state populations change with the delay time; the more effective scheme for controlling the population transfer of the three-level Na2 is the one with a short pulse width and positive delay time, which is explained by the light-induced potentials (LIPs). Hu et al.[22] and Yan et al.[23] studied the sensitivity of the population transfer and the angular distribution of population of the four-level Li2 to the molecular rotation. Han et al.[24] and Jing et al.[25] presented that the laser intensity, wavelength, and delay time affect the state population of the four-level Li2. Liu et al.[26,27] proposed that the delay time only alters the peaks height, but does not change the peak positions of the photoelectron spectra of Li2. Miao et al.[28,29] suggested that the peak positions and heights in the photoelectron spectra of 
The Autler–Townes (AT) triple splitting and the selective distributions of the dressed states may be attained by altering the laser intensity, wavelength, and pulse envelope.[30–32] No studies focus on the sensitivity of the wave packet dynamics of the four-level ladder K2 to the delay time. New data on the delay time dependence of the photoelectron spectra and state populations of the four-level ladder K2 molecule is obtained in the presence of two control pump fields and one probe field via the time-dependent quantum wave packet method.
2. Theory
Four states (ground state 

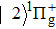

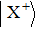
The wave functions of the three-state model are written in the column vector
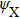


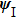
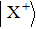

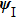

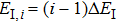



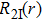











The split-operator Fourier method is used to solve the time-dependent Schrödinger equation.[29,30] The population on each electronic state is written as[2–13,22,24,25,27,29]

From the dressed-state theory, the excited state ∣2〉 is dressed by the laser field and splits into three substates ∣α〉, ∣β〉, and ∣γ〉 with energy 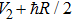

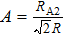



3. Results and discussion
The delay time dependence of the wave packet motion is studied. Figure 





 | Fig. 2. The evolution of the wave packet on excited states ∣A〉 and ∣2〉 with time and internuclear separation: (a) and (c) 
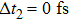


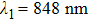



|
 | Fig. 3. The evolution of photoelectron spectra of K2 (a) at different delay time 
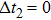


|
The delay time dependence of the photoelectron spectra is studied. Figure 
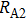




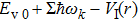
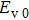
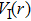



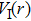




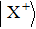

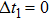
The delay time dependence of the state populations is studied. Figure 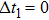



 | Fig. 4. (color online) The evolution of state populations (a)–(f) at different delay time 




|
4. Conclusions
The effect of the delay time (case 1: 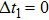



Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] | |
| [33] | |
| [34] | |
| [35] | |
| [36] | |
| [37] | |
| [38] | |
| [39] | |
| [40] |