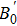
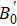
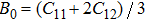
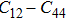
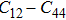
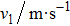
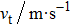
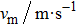
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Structural, electronic, elastic, and thermal properties of CaNiH3 perovskite obtained from first-principles calculations
Cite this Article
Benlamari S, Bendjeddou H, Boulechfar R, Amara Korba S, Meradji H, Ahmed R, Ghemid S, Khenata R, Bin Omran S. Structural, electronic, elastic, and thermal properties of CaNiH3 perovskite obtained from first-principles calculations. Chinese Physics B, 2018, 27(3): 037104 
Permissions
Structural, electronic, elastic, and thermal properties of CaNiH3 perovskite obtained from first-principles calculations
† Corresponding author. E-mail:
Abstract
Abstract
A theoretical study of the structural, elastic, electronic, mechanical, and thermal properties of the perovskite-type hydride CaNiH3 is presented. This study is carried out via first-principles full potential (FP) linearized augmented plane wave plus local orbital (LAPW+lo) method designed within the density functional theory (DFT). To treat the exchange–correlation energy/potential for the total energy calculations, the local density approximation (LDA) of Perdew–Wang (PW) and the generalized gradient approximation (GGA) of Perdew–Burke–Ernzerhof (PBE) are used. The three independent elastic constants (C11, C12, and C44) are calculated from the direct computation of the stresses generated by small strains. Besides, we report the variation of the elastic constants as a function of pressure as well. From the calculated elastic constants, the mechanical character of CaNiH3 is predicted. Pertaining to the thermal properties, the Debye temperature is estimated from the average sound velocity. To further comprehend this compound, the quasi-harmonic Debye model is used to analyze the thermal properties. From the calculations, we find that the obtained results of the lattice constant (a0), bulk modulus (B0), and its pressure derivative (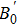
1. Introduction
In recent years, ternary hydrides ABH3 have attracted considerable attention of a lot of scientists around the world because of their high hydrogen storage capacity and large number of other applications like switchable mirrors,[1–3] rechargeable batteries, energy storage, etc.[4,5] In ABH3 ternary hydrides, A is usually an alkali or alkaline earth metal and B is a transition metal. Some of the ABH3 ternary hydrides also show the perovskite-type structure in which H is regarded as the anion, and A and B are monovalent and divalent cations, respectively. If A is a heavier alkaline metal like Ca, Ba, Sr and B is Fe, Ni, or Co, the resultant ABH3 compounds are reported stable, and this stability is associated with the filled d-band of the alkaline earth and transition metals. However, the priority to these hydrides is given mainly on the basis of their good storage capacities, good reversibility, and large reactivity.
The CaNiH3 based alloys are expected to be attractive hydrogen storage materials in the coming hydrogen energy era because of their good hydrogenation properties and relatively low material cost. The perovskite-type hydride CaNiH3 was discovered in the study of the decomposition of hydrogenated CaNiH3 alloys in the H2 atmosphere.[6] This ternary hydride has a large hydrogen content of 3 wt.% and decomposes to CaH2 and Ni when it is heated from room temperature to 773 K in an H2 atmosphere of 3 MPa. Some investigations have been reported in the literature.[6–9] The first work on the hydrogenation properties of CaNiH3 was reported by Oesterreicher et al.[7] More recently, Takeshita et al.[6] reported the synthesis and decomposition of CaNiH3 hydride at high temperatures up to 773 K, analyzed the hydrogenation properties and the change in crystal structure of CaNiH3 by using differential thermal analysis (DTA) and x-ray diffraction (XRD). Sato et al. calculated its electronic band structures by employing the augmented plane wave method[8] and predicted the metallic nature of the CaNiH3 compound. An experimental study of the formation and thermal desorption process of CaNiH3 was also carried out.[9]
Although studies are reported on the hydrogenation/dehydrogenation processing, to the best of our knowledge, there are no experimental or theoretical data on the elastic, mechanical, and thermal properties of this interested compound, which are important to understand the real character of CaNiH3 and its applicability for different applications. Thus the aim of this work is to give a comprehensive study on its physical properties by employing full potential (FP) linearized augmented plane wave plus local orbital (LAPW+lo) method framed within density functional theory (DFT) and implemented in the WIEN2k computational package. The organization of the rest of the paper is as follows. In Section
2. Calculation method
The first-principles calculations of this work are performed using the FP-LAPW+lo method[10] based on the DFT[11] as implemented in WIEN2k code.[12] In this approach, the crystal unit is partitioned into two regions: nonoverlapping muffin tin spheres (MT) and interstitial regions (IR). In the two regions, the Kohn Sham wave functions, charge density, and potential are treated differently. To determine the structural parameters, the local density approximation (LDA) of Perdew–Wang (PW)[13] and the generalized gradient approximation (GGA) of Perdew–Burke–Ernzerhof (PBE)[14] are employed. To avoid the overlapping, the muffin-tin radii (RMT) are taken to be 2.5, 2.03, and 1.10 for Ca, Ni, and H, respectively. To expand the charge density as well as the potential, the maximum angular moment 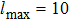
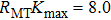
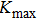
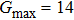
3. Results and discussion
3.1. Structural properties
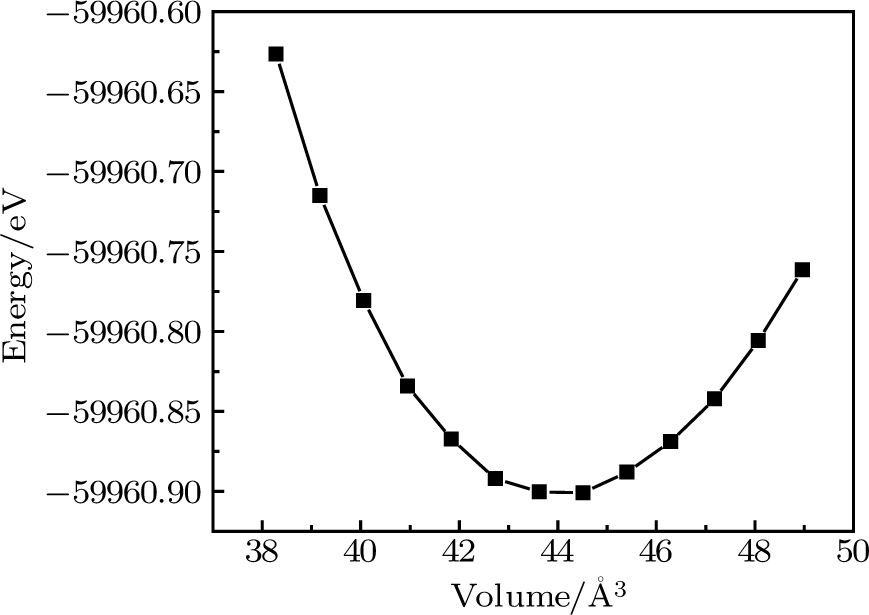
The CaNiH3 ternary hydride, typically containing a single molecule, crystallizes in the perovskite-type cubic structure with 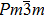
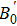
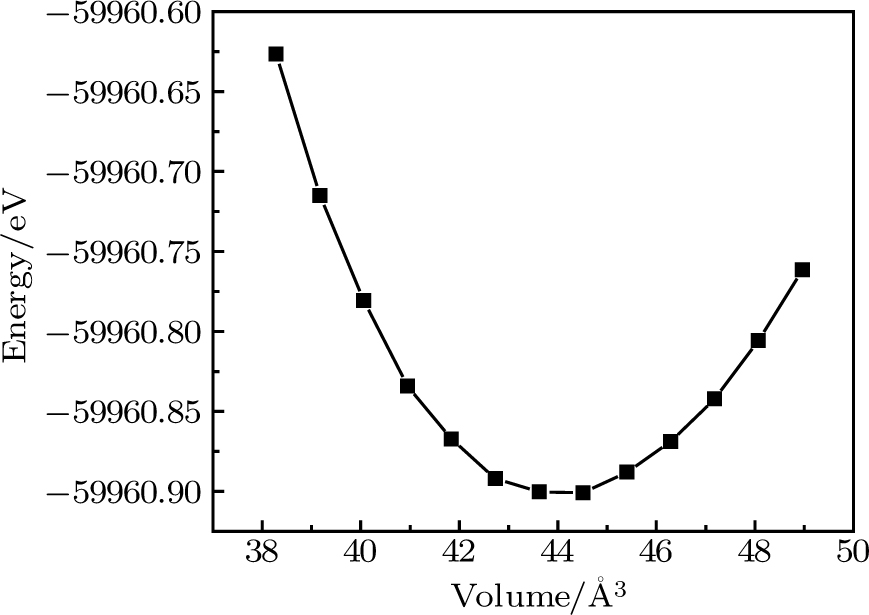 | Fig. 1. Calculated total energy versus volume for CaNiH3 using GGA approximation. |
| Table 1.
Calculated lattice constant a0, bulk modulus B0, its pressure derivative |
3.2. Elastic properties
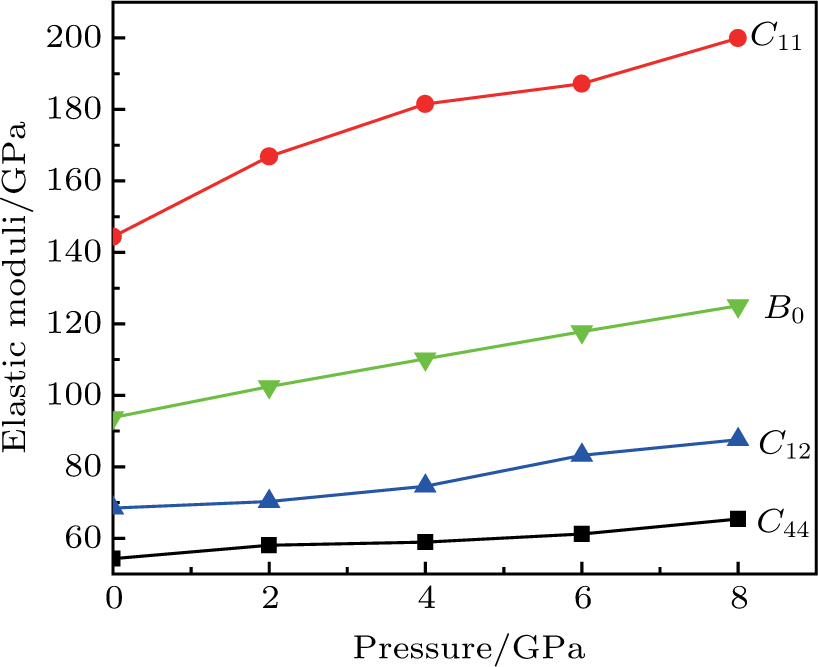
The study of the elastic parameters is crucial to getting knowledge about the bonding among the nearest neighbors, structural stability, and anisotropic character of a material, and these parameters are vital to understanding the mechanical behavior. Moreover, studying the aforementioned parameters at the high pressure also provides information about the material response, mechanical stability, and strength under compression. As our system is cubic, it is well recognized that the three independent elastic constants are C11, C12, and C44. To calculate the elastic constants, the Thomas Charpin method as integrated within the WIEN2k package is used[12] and the obtained elastic constants for the CaNiH3 compound are given in Table 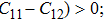
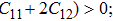
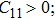
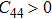
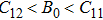
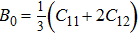
3.3. Mechanical properties
Knowledge of the elastic properties of materials promotes the understanding of the fundamental aspects of the mechanical deformation and structural properties of a crystal. In order to gain a deeper insight into the mechanical properties of the material, we calculate some polycrystalline elastic moduli such as the anisotropy factor A, Youngʼs modulus E, Poissonʼs ratio υ, and shear modulus G by employing the Voigt Reus Hill approach and expressions given in the followings:[18–21]
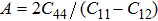
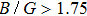
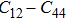
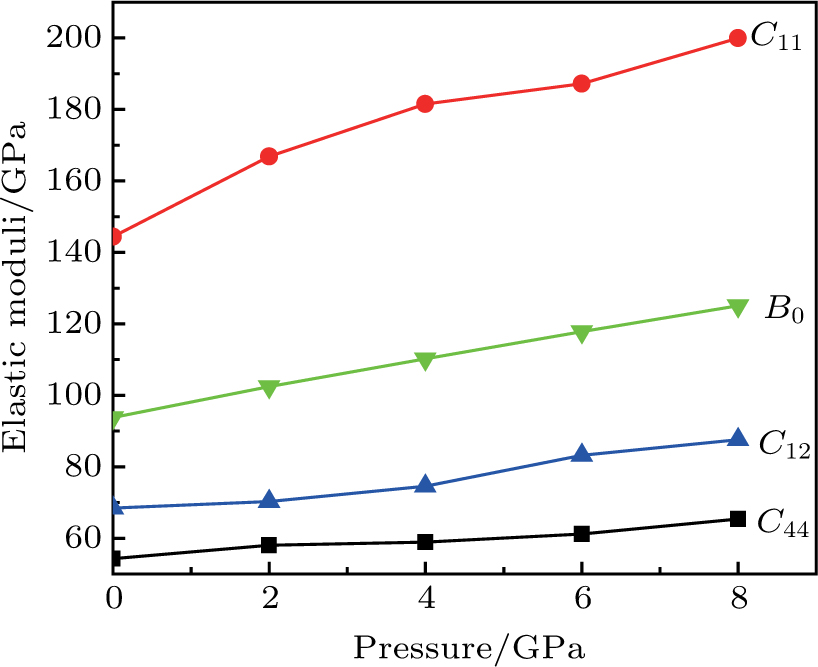 | Fig. 2. (color online) Calculated pressure dependence of elastic constants Cij and bulk modulus B0 for CaNiH3. |
| Table 2.
Calculated density ρ, anisotropy factor A, Youngʼs modulus E, Poissonʼs ratio υ, shear modulus G, (B/G) ratio, and Cauchyʼs pressure |
3.4. Debye temperature calculation
The role of the Debye temperature in the lattice vibration theory is the same as that of the Fermi temperature in the theory of electrons of metals since both represent a measure of the temperature to separate the low-temperature region (where quantum statistics is used) from the high temperature region (where classical statistics is found to be applicable). This is a very noteworthy parameter which is coupled to many materials’ thermal characteristics, in turn correlating to a lot of their physical properties, for instance, elastic constants, specific heat, and melting temperature. Therefore, the knowledge of this parameter is essential for suggesting a material suitability for the electronic devices.
To calculate the Debye temperature θD, a typical technique is derived from the data of elastic constants in which θD is estimated by averaging sound velocity vm by employing the equation[28]
To calculate the average sound velocity, the following relation is used:
| Table 3.
Calculated longitudinal vl, transverse vt, average elastic wave velocities vm, and Debye temperature θD for CaNiH3. . |
3.5. Electronic properties
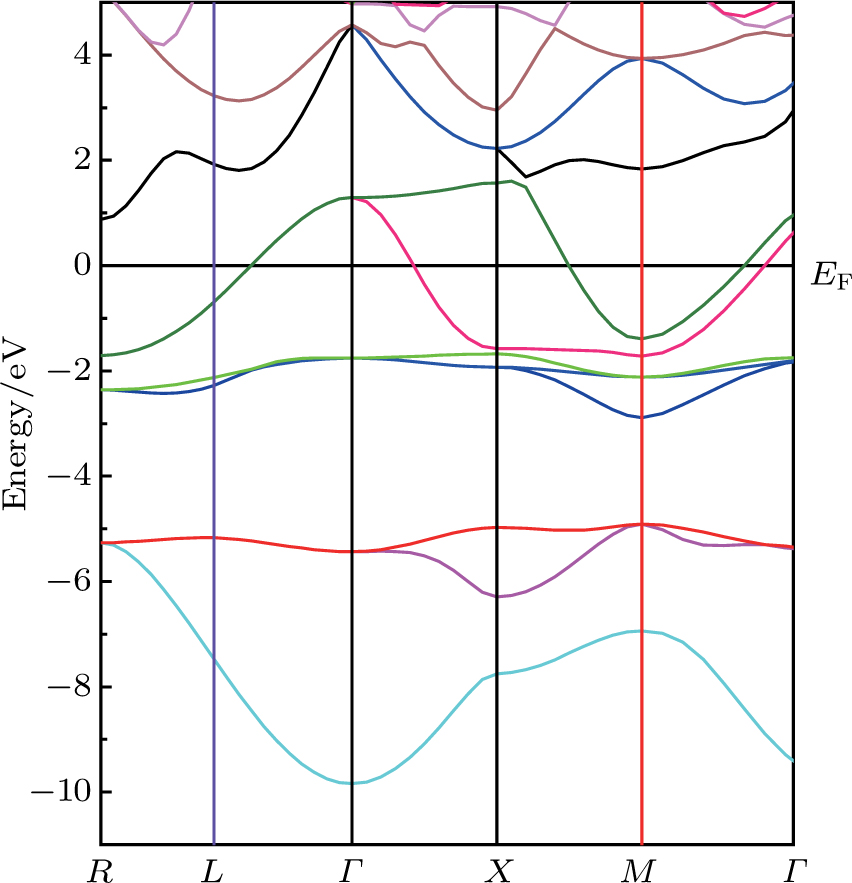
The calculated band structure of the perovskite-type hydride CaNiH3 along the higher symmetry directions in the Brillouin zone using (PBE-GGA) approach is shown in Fig.
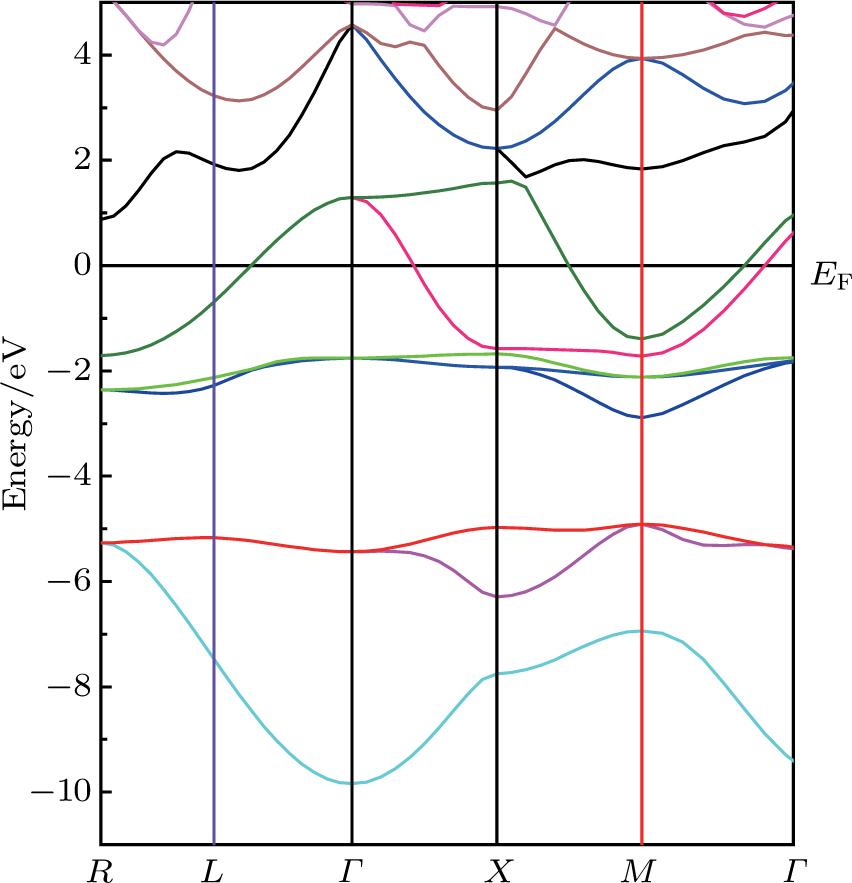 | Fig. 3. (color online) Band structure alone high-symmetry directions in the Brillouin zone for the CaNiH3 compound. |
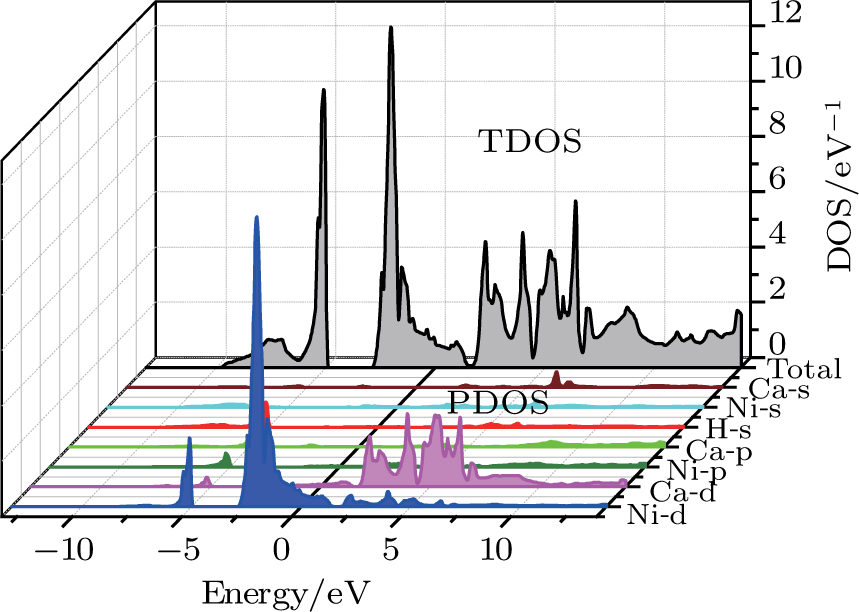
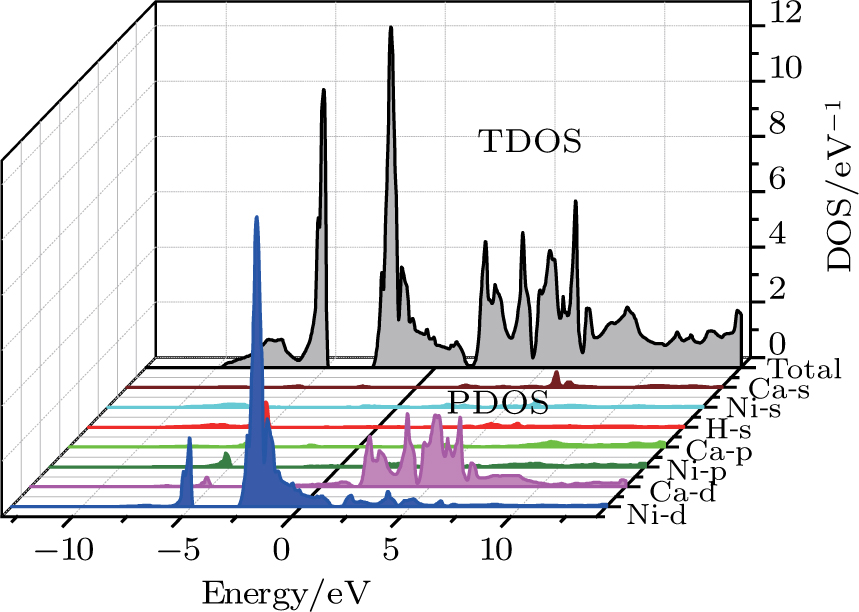 | Fig. 4. (color online) Calculated total and partial densities of states for the CaNiH3 compound. |
3.6. Thermal properties
In order to determine the thermal effects at high pressure and high temperature, Gibbs computational code based on the quasi-harmonic Debye model is used.[30] In this method, Gibbs function 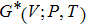
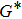
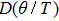
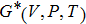
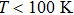
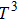
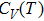
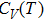
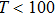
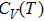
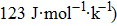
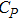
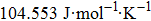
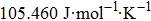
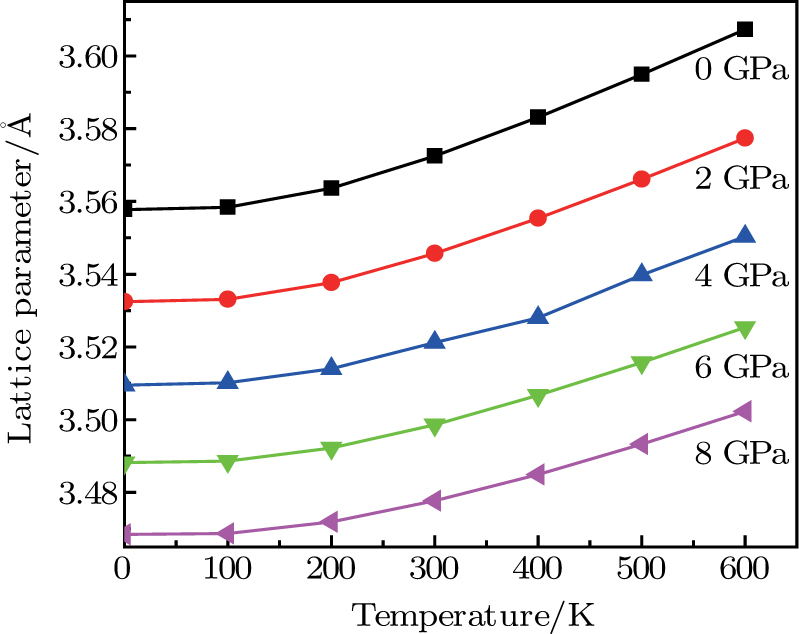
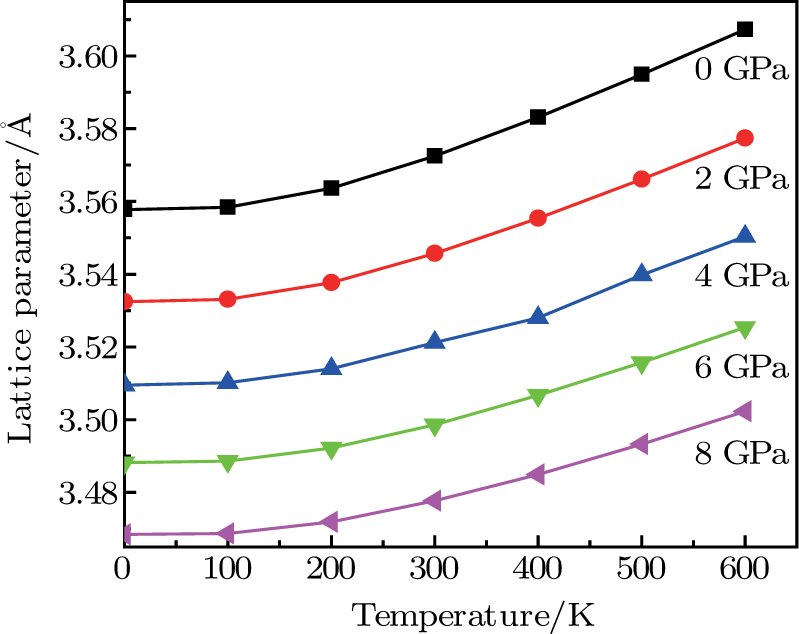 | Fig. 5. (color online) Variation of the lattice constant as a function of temperature at different pressures for CaNiH3. |
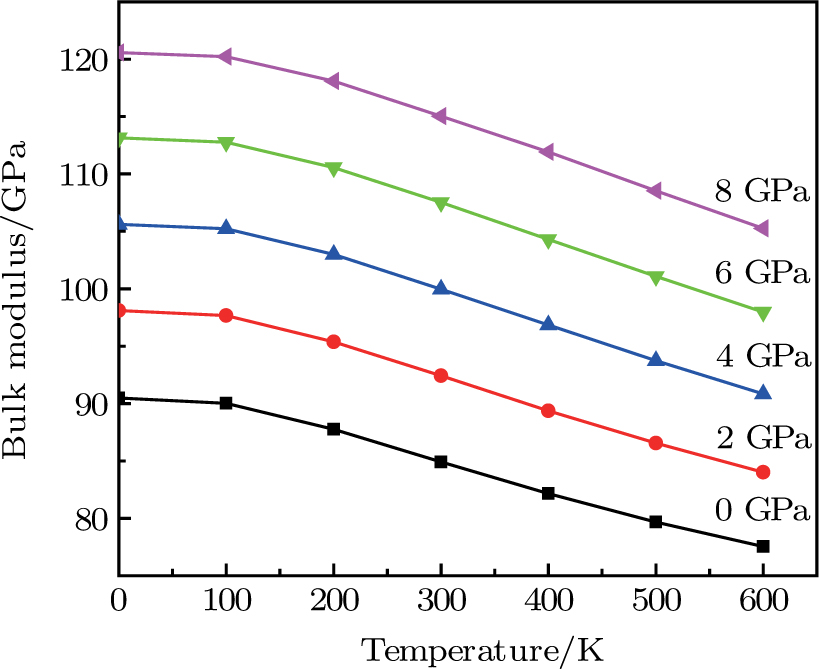
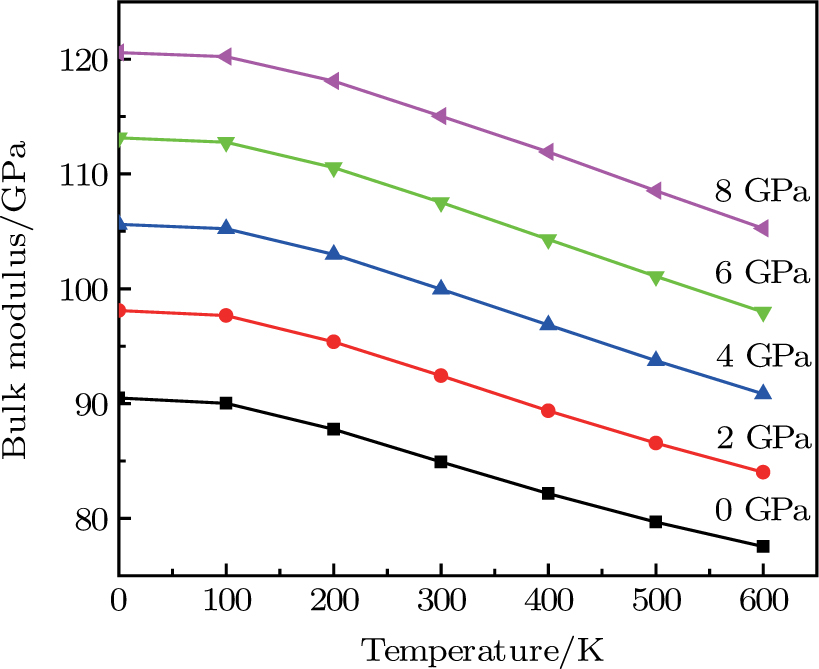 | Fig. 6. (color online) Variation of the bulk modulus as a function of temperature at different pressures for CaNiH3. |
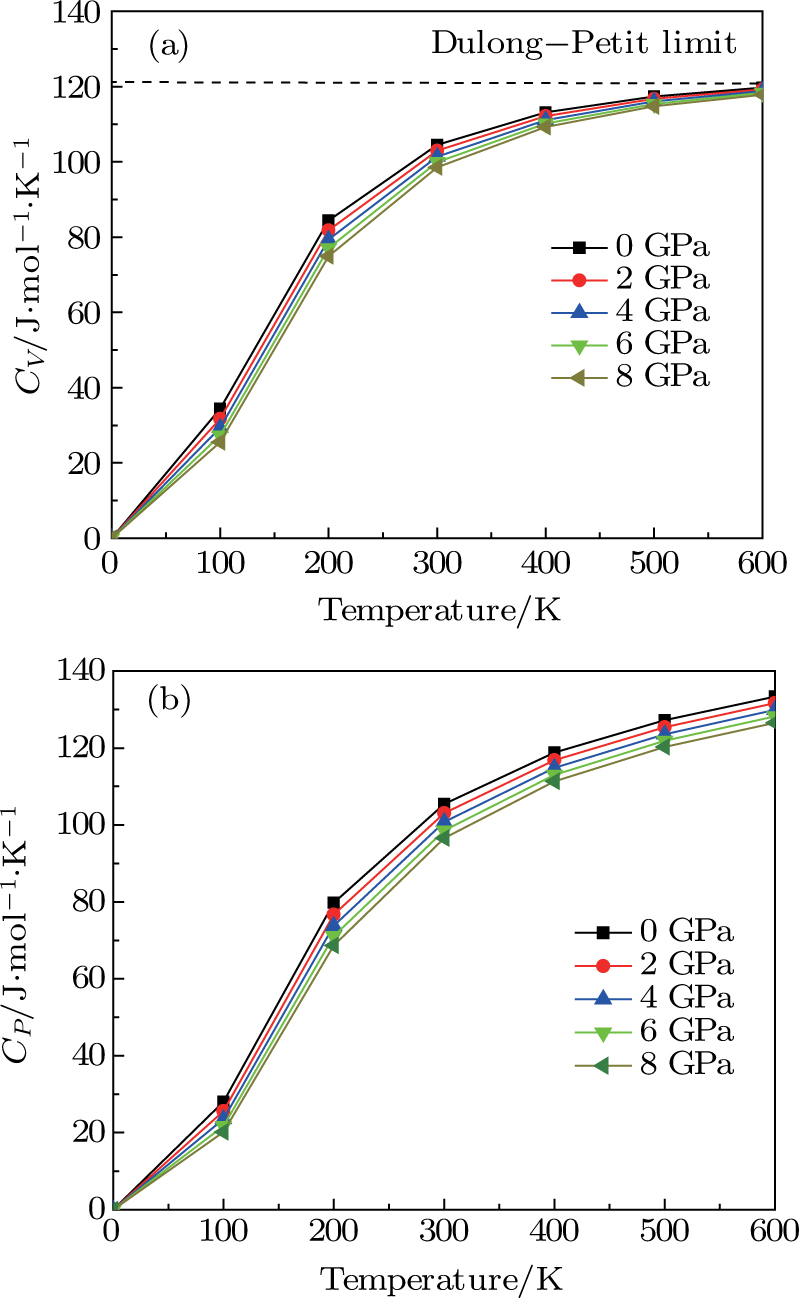
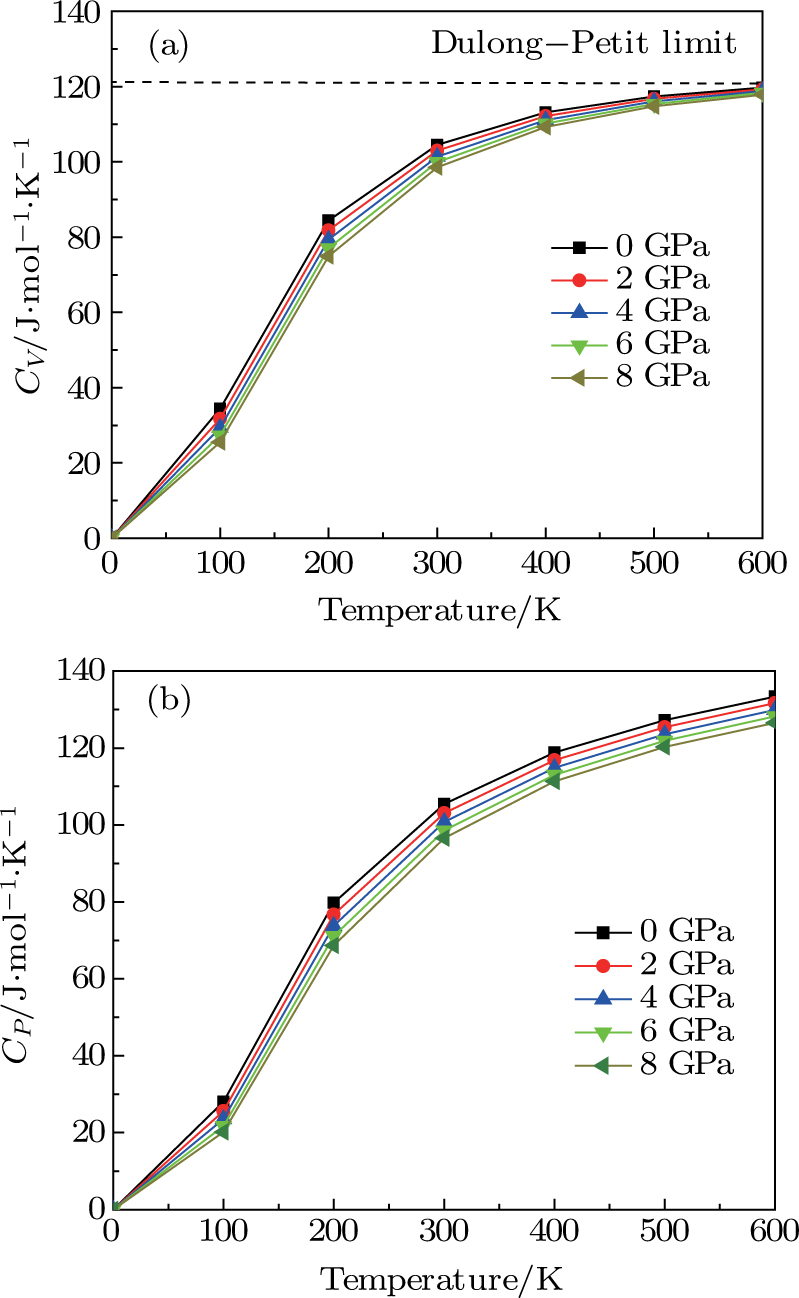 | Fig. 7. (color online) Variation of the heat capacities (a) CV and (b) 
|
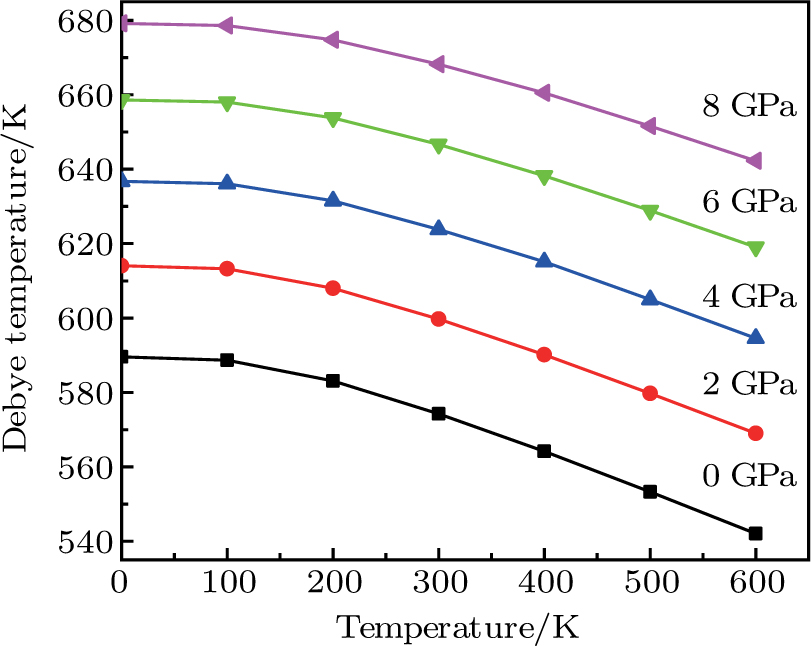
The variation of the Debye temperature with respect to the pressure is presented in Fig.
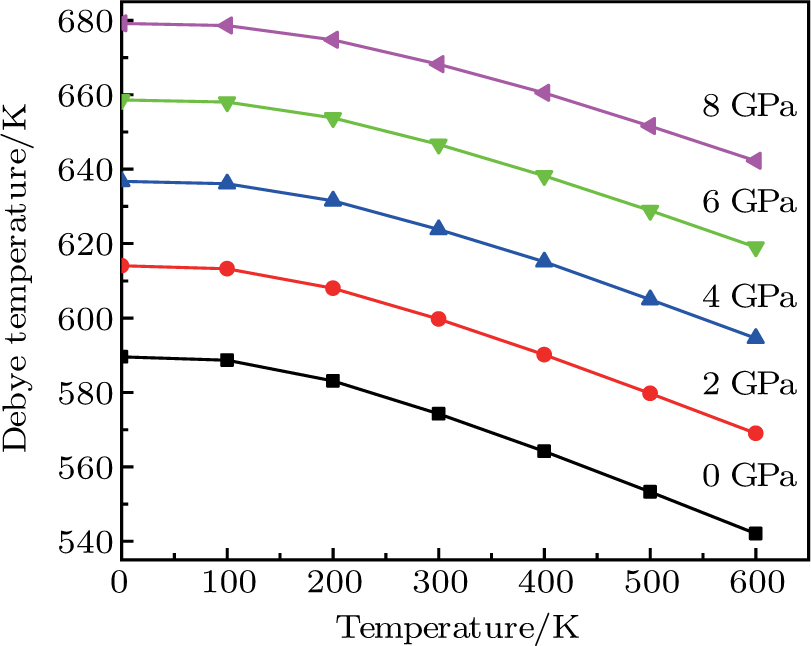 | Fig. 8. (color online) Variation of the Debye temperature θD with the temperature at different pressures for CaNiH3. |
4. Conclusion
The calculations of the structural, elastic, thermal, and electronic properties of the CaNiH3 ternary compound were carried out by using ab initio FP-LAPW+lo computational method at the level of PW-LDA and PBE-GGA exchange–correlation energy functional. We found that the obtained results of the lattice constants, as well as bulk modulus, were in a nice agreement with the available data in the literature. Also, we have computed elastic constants C11, C12, and C44 to determine the shear modulus, anisotropy factor, and Debye temperature. It was found that our computed elastic constants of CaNiH3 adequately followed the stability criteria whereas its electronic band structure and DOS profile demonstrated its metallic nature. To understand the dependence of the lattice parameters, bulk modulus, Debye temperature, and specific heat capacity on the temperature and pressure, the quasi-harmonic Debye model was employed. It was deduced that our predictions concerning specific heat CV of the CaNiH3 hydride were closely following the Dulong–Petit law, showing that our results were in line with many other solids at high temperature. Hence, our first-principles DFT calculations performed in this study can be a very useful as a guide to complement future experiments.
Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] | |
| [33] | |
| [34] | |
| [35] | |
| [36] | |
| [37] | |
| [38] |