|
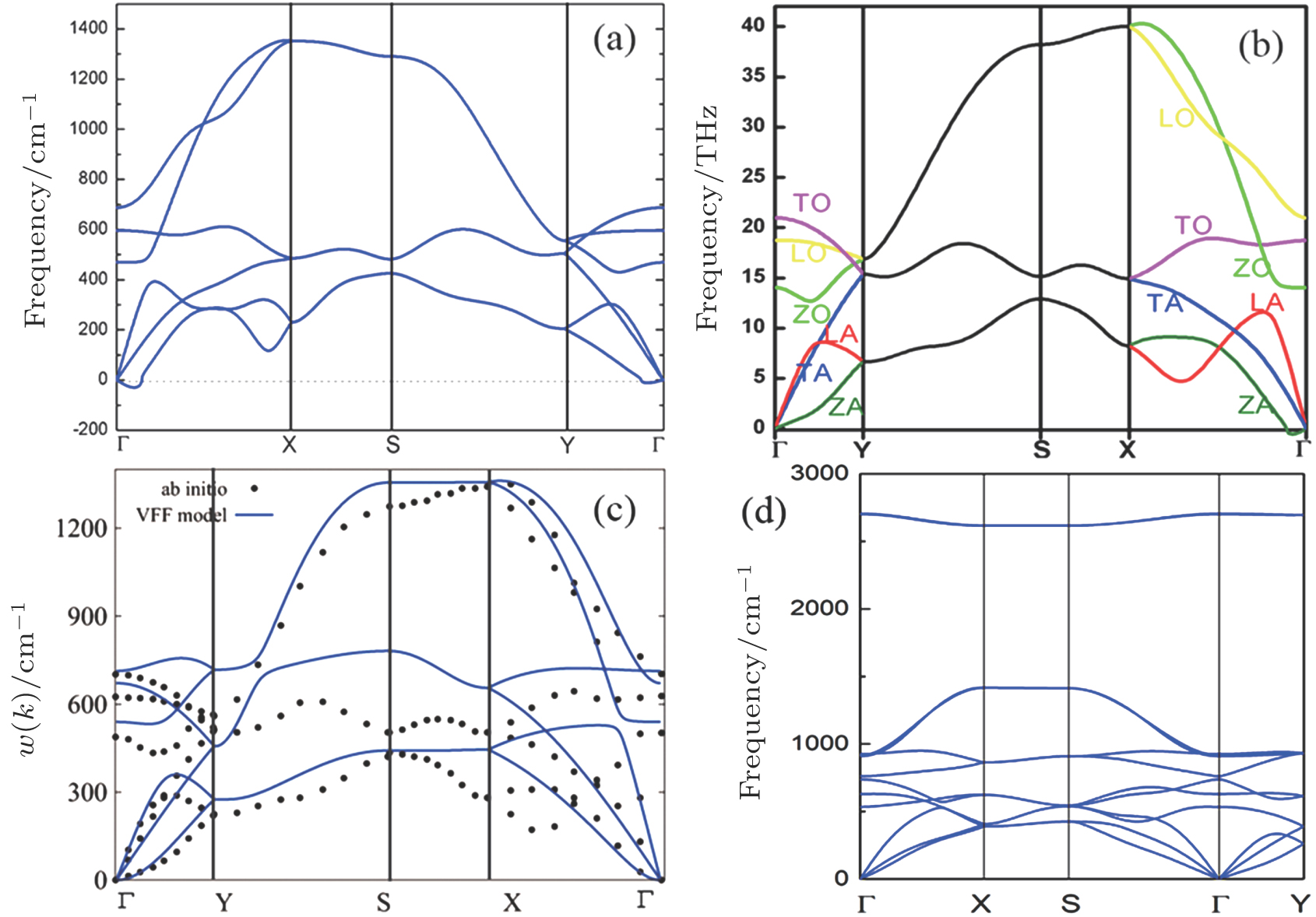
(color online) Phonon spectra of δ6 borophene calculated by (a) density function perturbation theory (DFPT) and (b) finite displacement (FDS) method, adapted with permission form Refs. [53] and [54], respectively. (c) Phonon spectrum calculated by molecular dynamics simulations within valence force filed (VFF) mode, compared to the result of ab-initio calculation, with permission from Ref. [57]. (d) The phonon spectrum of fully hydrogenated borophene (borophane), adapted from Ref. [58].
|