











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Excited state intramolecular proton transfer mechanism of o-hydroxynaphthyl phenanthroimidazole
Cite this Article
Liu Shuang, Ma Yan-Zhen, Yang Yun-Fan, Liu Song-Song, Li Yong-Qing, Song Yu-Zhi. Excited state intramolecular proton transfer mechanism of o-hydroxynaphthyl phenanthroimidazole. Chinese Physics B, 2018, 27(2): 023103 
Permissions
Excited state intramolecular proton transfer mechanism of o-hydroxynaphthyl phenanthroimidazole
† Corresponding author. E-mail:
Abstract
By utilizing the density functional theory (DFT) and the time-dependent density functional theory (TDDFT), the excited state intramolecular proton transfer (ESIPT) mechanism of o-hydroxynaphthyl phenanthroimidazole (HNPI) is studied in detail. Upon photo is excited, the intramolecular hydrogen bond is obviously enhanced in the S1 state, which thus promotes the ESIPT process. Hydrogen bond is shown to be strengthened via comparing the molecular structures and the infrared vibration spectra of the S0 and S1 states. Through analyzing the frontier molecular orbitals, we can conclude that the excitation is a type of the intramolecular charge transfer excitation, which also indicates the trend of proton transfer in S1 state. The vertical excitation based on TDDFT calculation can effectively repeat the absorption and fluorescence spectra of the experiment. However, the fluorescence spectrum of normal structure, which is similar to the spectrum of isomer structure is not detected in the experiment. It can be concluded that the fluorescence measured in the experiment is attributed to both structures. In addition, by analyzing the potential energy curves (PECs) calculated by the B3LYP functional method, it can be derived that since the molecule to cross the potential barrier in the S1 state is smaller than in the S0 state and the reverse proton transfer process in the S1 state is more difficult than in the S0 state, the ESIPT occurs in the S1 state.
1. Introduction
As one of the cases of most important weak interactions, hydrogen bond plays quite a vital role in the study of photochemistry, such as supramolecular photochemistry, material photochemistry, biochemistry, and photochemistry of solution, etc.[1–5] Since the pioneering work by Weller et al.,[6,7] the excited state intra- and inter-molecular proton transfer (ESIPT) have become a research focus in experiment and in theory.[1,2,8–11] For example, Zhao et al. have been deciphered the excited state behavior for 2- (
Since the phenanthroimidazole derivatives have good molecular structural characteristics, they have aroused the great research interest of researchers. By using the TiO2(R) nano-semiconductor as the catalyst under solventless condition, Jayabharathi et al.[29] synthesized five phenanthroimidazole derivatives, which were then characterized via nuclear magnetic resonance (NMR) and single crystal x-ray diffraction (XRD) techniques. Shahid and Misra[30] synthesized three molecular systems based phenanthroimidazole moiety and studied photo enolization via ESIPT mechanism. The results revealed that the o-hydroxynaphthyl phenanthroimidazole (HNPI) and o-hydroxyphenyl phenanthroimidazole (HPPI) show light triggered enolization. However, benzyl phenanthroimidazole (BPNI) could not show the phenomenon of proton transfer due to the absence of OH moiety. Specifically, fluorescence “turn-off” and “turn-on” behavior could occur when HNPI interacts with AcO−, which thus could be used as a probe of AcO−.
Although Shahid and Misra[30] experimentally studied in detail the photo enolization process of HNPI triggered by the ESIPT mechanism, they obtained only the geometry optimization and frontier molecular orbital by quantum chemical calculation, which cannot be quantitatively nor qualitatively explain the ESIPT mechanism. Thus we are motivated to carry out a clear and detailed theoretical investigation on ESIPT of HNPI by utilizing DFT and TDDFT methods.[31] We study the excited state dynamical process of HNPI by performing detailed calculation on the properties of both the ground and excited states, such as the geometry optimization, potential energy curves, the frontier molecular orbital, reduced density gradient (RDG), IR spectrum, absorption and emission spectra, etc. We investigate the ESIPT mechanism of HNPI in detail, which is not only to clarify excited state dynamical process of HNPI system, but also to facilitate the understanding of intramolecular hydrogen bond dynamical processes.
The rest of this paper is organized as follows. In Section
2. Computational details
The S0 and S1 state optimizations of HNPI molecule are carried out by using the DFT and TDDFT methods, which are implemented in the Gaussian 09 program suite.[31] B3LYP[32] functional and 6-31+G (d) basis set[33] are employed in both the DFT and TDDFT calculations. In order to simulate the environment of the acetonitrile solvent, the polarizable continuum model (PCM) and integral equation formalism variant PCM (IEFPCM)[34–37] are employed in the present work, which can reproduce the experimental results well. All the bond lengths, bond angles and dihedral angles are relaxed in the optimization of HNPI molecule in S0 and S1 states. In order to verify the structures corresponding to local minima in S0 and S1 states, the vibrational frequencies are calculated, which give no imaginary frequency. By using the optimized geometry of ground-state HNPI molecule, the vertical excitations are obtained by TDDFT calculation considering ten low-lying electronic states.[1,16,18,38–42] The PECs of S0 and S1 states are scanned,[43] which follows a constrained optimization by varying O–H bond length in the enol form ranging from 0.9 Å to 1.9 Å in steps of 0.03 Å. The vibration frequencies are determined via the diagonalization of the Hessian matrix. The gradients of dipole moment are utilized to determine the infrared (IR) intensity. In order to distinguish among the different types of interactions in HNPI molecule, the reduced density gradient (RDG) is calculated using Multiwfn.[44] Such as RDG is then visualized, from which the hydrogen bond can be easily recognized.
3. Results and discussion
3.1. Optimization of HNPI
The enol and keto forms are optimized employing DFT and TDDFT methods in ground (S0) and excited (S1) states of HNPI, respectively, which is subsequently followed by vibrational frequency analysis in order to validate such stationary points. The optimized enol and keto structures in ground state are shown in Fig.
 | Fig. 1. (color online) Optimized structures of HNPI molecules of enol and keto forms in S0 state: blue: H, yellow: C, red: O, pink: N. The dash line refers to the intramolecular hydrogen bond. |
| Table 1.
Primary optimized bond lengths (in unit Å) and bond angles (°) of hydrogen bond groups for normal form (enol) and tautomeric forms (keto) of the HNPI in S0 and S1 states. . |
A clear-cut feature of hydrogen bond dynamics can be obtained by analyzing the vibrational frequency of O1–H2 moiety which is related to the O1–H2 ⋯ N3. It is well known that IR spectrum is an effective method to determine whether the proton transfer occurs, which is judged by the blue shift or red shift of the IR spectrum.[45] Thus, the IR spectra of HPNI in enol form are calculated both for the S0 state and for the S1 state, which is shown in Fig.
 | Fig. 2. (color online) IR spectra of enol-form HPNI at the spectral region of the O1–H2 stretching band in the S0 and S1 states. |
3.2. Frontier molecular orbital (MO) analyses
The frontier molecular orbitals (mainly the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO)) are calculated and analyzed for better understanding the charge distribution and charge transfer in the HNPI molecule. In Fig. 
 | Fig. 3. (color online) Visual electron population of the frontier molecular orbitals HOMO and LUMO. |
| Table 2.
Excitation wavelengths, oscillator strengths and the corresponding orbital transition contributions of HNPI molecule. . |
3.3. Absorption and emission spectra
The absorption and fluorescence spectra of HNPI molecule are displayed in Fig.
 | Fig. 4. (color online) Absorption (abs) and fluorescence (flu.) spectra of HNPI, obtained at the calculated level of B3LYP/6-31+G (d)/IEF-PCM (acetonitrile). |
3.4. Potential energy curves
For a more comprehensive understanding of the process that the proton migrates from the hydroxyl group to nitrogen atom, we plot in Fig.
 | Fig. 5. (color online) Curves of potential energy versus O1–H2 bondlength in a frequency range from 0.9 Å to 1.9 Å of the HNPI molecule in S0 and S1 states. |
3.5. Discriminating weak interaction types by filling color to RDG isosurfaces
where RDG is the reduced density gradient and
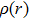
is the total electron density. According to atom in molecule (AIM) theory,[50] the relationship between the second largest eigenvalue λ2 and
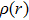
of the electron density Hessian matrix as shown in Eq. (2 ),
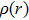
is an important measure of weak interaction intensity, while the type of weak interaction is represented by the sign (
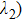
function. Therefore, the weak interactions of different types can be analyzed by drawing the scatter graph of function 1 (RDG) and function 2 (
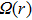
). The function obtained by multiplying the function of
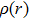
and sign (
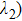
and thus yielding the RDG isosurface is shown in Fig. 6(a) with a contour value of 0.5 and the RDG isosurface ranging from −0.04 to 0.02. The spike of the hydrogen bond is located at −0.036 a.u., which indicates that the hydrogen bond is extremely strong in the S1 state. Similarly, the weak interactions can be distinguished in Fig. 6(b) . It can be explicitly observed from Fig. 6(b) that there is a strong hydrogen bond between
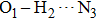
.
By combining the RDG function and the 
 |
 |
 | Fig. 6. (color online) (a) Plot of reduced density gradient (RDG) versus 
|
4. Conclusions and perspectives
The DFT/TDDFT/B3LYP functional method is utilized to study the ESIPT mechanism of HNPI molecule. We can conclude that the proton transfer occurs in the S1 state via analyzing the calculated electron spectrum, infrared spectrum and potential energy curves. In addition, by analyzing the bond lengths, bond angles and the frontier molecular orbitals in the S0 state and the S1 state, respectively, we can prove that the hydrogen bond interaction can be strengthened, which plays a decisive role in proton transfer. In addition, we carry out a detailed study of the proton transfer process of HNPI molecule by calculating the PECs of S0 and S1 states. The obtained potential barriers of reversed proton transfer are 4.28 kcal/mol in the S1 state and 2.2 kcal/mol in the S0 state, respectively. Thus it can be concluded that the reverse proton transfer process in the S0 state is more difficult than in the S1 state. Therefore, we can derive from the above analysis that the proton transfer occurs in the S1 state, which is of great significance for doing molecular research.
Reference