



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Influence of the initial electronic state on minima of high-order harmonic spectrum radiated from hydrogen molecular ion
Cite this Article
Cui Hui-Fang, Miao Xiang-Yang. Influence of the initial electronic state on minima of high-order harmonic spectrum radiated from hydrogen molecular ion. Chinese Physics B, 2017, 26(10): 103302 
Permissions
Influence of the initial electronic state on minima of high-order harmonic spectrum radiated from hydrogen molecular ion
† Corresponding author. E-mail:
Project supported by the National Natural Science Foundation of China (Grant No. 11404204) and the Program for the Top Young Academic Leaders of Higher Learning Institutions of Shanxi Province, China.
Abstract
We theoretically investigate the high-order harmonic generation of the one-dimensional hydrogen molecular ion at fixed intermediate internuclear distance, driven by a multicycle laser field. Our results show that the initial electronic state of the hydrogen molecular ion affects the modulation of the high-order harmonic spectrum, especially the positions of the minima. Based on the two-state model, the underlying physical mechanism of the minimum is analyzed and discussed. Further analysis shows that the different positions of the minima in the different initial electronic states can be understood via the different interferences of the two phase-adiabatic states at the ionization times.
1. Introduction
Recently, high-order harmonic generation (HHG) and its application have continuously been investigated as it can be used to produce attosecond laser pulses, and to probe the structure[1–3] and electronic dynamics[4–6] of atoms and molecules. The mechanism of HHG from atoms is described by the semiclassical three-step model:[7] First, the electron escapes from the core by tunnelling through the Coulomb potential barrier, which is distorted by the intense laser field. Second, the electron is accelerated away from its parent ion by the laser field and obtains additional kinetic energy. Finally, the electron recombines with the parent ion and emits HHG photons. Based on the conventional model of strong-field ionization, the electron will leave the atom with the largest probability at the peaks of the laser field. However, the electron dynamics in molecules is qualitatively different from that in atoms due to the more complicated structures of molecules.[8–14] As the simplest molecule, the hydrogen molecular ion (

The importance of the electronic structure of the molecule in molecular HHG has been pointed out in the previous theoretical and experimental studies.[29–33] In this study, we first investigate the role of the electronic state in HHG minimum from 
2. Theoretical method
When we consider the linearly polarized laser field along the molecular axis, the Hamiltonian of this system (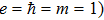
where
denotes the electronic kinetic-energy operator, 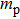
where R is the internuclear distance of the molecular ion.[36] The external interaction between the laser field and the molecule is given by
where ω0 is the angular frequency and 
where T is the pulse duration and E0 is the peak strength.
where T is the kinetic energy operator as in Eq. (2 ), and V is the interaction potential taking all the potential energy of the system plus a purely imaginary term to produce an absorbing boundary. In this study, we set R = 7 a.u. The ground and the first excited state energies of this model are 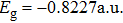
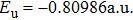
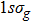
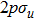
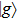
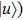
1 , the two lowest-lying states have opposite parities and their superpositions form two localized states 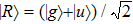
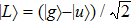
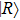

All the calculations were performed for the 1D fixed-nuclei model of 
 |
 |
 |
 |
 |
The electric field of the laser pulse is assumed to have the following form:
 |
 |
Equation (
 |
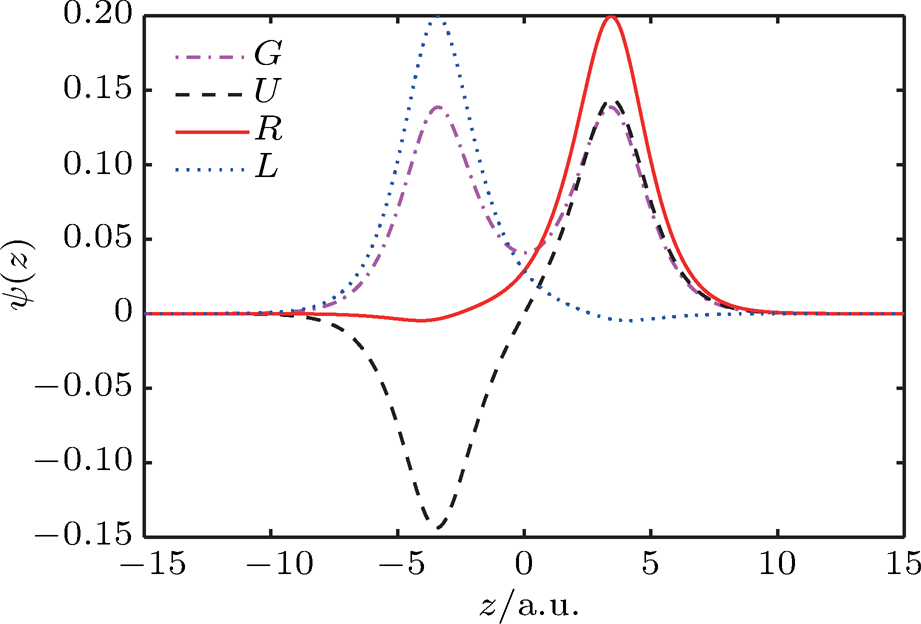 | Fig. 1. (color online) Plots of ψ–z of electronic eigenstates 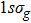 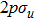 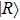 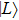 |
3. Results and discussion
where 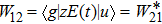
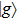
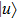
where
with the BO energy separation 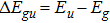
The two eigenfunctions 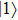
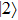
where the time-dependent wave function 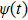
7 ), 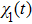
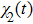
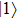
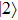
where 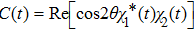
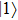
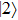
The time-dependent local populations 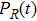
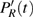
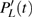
3(a) . From this figure, we note that 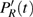


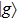

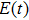

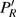
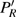
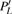
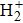
3(b) and 3(c) show the local population 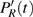
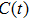
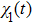
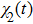
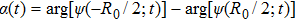
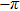
3(d) and 3(e) , the time-dependent phase difference 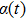
3(d) and 3(e) ) with the local populations (Figs. 3(b) and 3(c) ), we note that the counter-intuitive motion of the electron occurs when the phase difference passes through 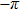
where 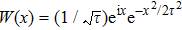
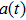
4(a) and 4(b) show the time-frequency distributions corresponding to the G case and U case, respectively. We focus on the ionization times between 819 a.u. and 916 a.u. In the G case, in each half cycle, there are two suppressed harmonic emissions denoted as G1 and G2 on the short quantum path, which match the spectral positions of the minima presented in Fig. 2 . There is also one suppressed harmonic emission labeled 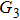
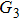
4(a) . In the U case, there are two obvious suppressed harmonic emissions denoted as U1 and 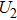
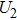
5 ). The electron is assumed to be a negative point charge, and is released from one of the protons of the molecule with 
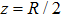
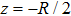
4(c) ]. For the 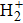
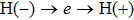
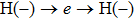
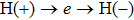
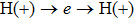
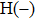
4(c) . To characterize the interference dynamics, figures 4(d) and 4(e) present the variations of the phase difference α at the time of ionization in the G case and in the U case, respectively. In the G case, during the ionization times of 819 a.u.–916 a.u., there are three instants at which 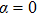
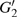
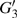
4(d) . As shown in Fig. 4(c) , the electron ionized from H(−) around 849 a.u. (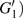

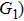
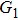
4(a) . The electron ionized from H(+) around 887 a.u. (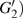
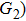
4(a) . The electron ionized around 868.5 a.u. (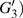
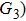

4(a) . In the U case, there are also three instants at which α = 0, and they are marked in the Fig. 4(c) as 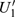
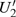
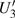
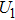
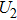

4(a) and Fig. 4(c) , respectively. The electron ionized from H(+) or H(−) around 900 a.u. recombines with one of the protons and emits the 60th harmonics around 972 a.u. which is in accordance with the suppressed harmonic emission 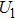
4(b) . At the instants of 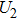
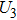
4(b) ], but they are not obviously linked to the spectrum minima due to the weaker intensity of the corresponding quantum paths. Therefore the suppressed harmonic emission is related to the moment of the counter-intuitive motion of electron and the mechanism of the HHG minimum is the consequence of the interference between the two phase-adiabatic states at the ionization time.
Figure 

 | Fig. 2. (color online) High-order harmonic spectra of   |
Due to the strong coupling between 

 |
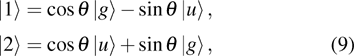 |
 |
 |
Using the two-phase-adiabatic state model, the total wave function is expressed as
 |
By projecting the wave function 


 |
 |
 |
 |
 | Fig. 3. (color online) (a) Time-dependent local population on the right proton, calculated by two-state model PR (blue dashed line), by numerical integration    |
In order to verify the viewpoint, we perform the time-frequency analysis by means of the wavelet transform,[40]
 |
 | Fig. 4. (color online) ((a) and (b)) Time-frequency distributions of the electron dipole acceleration throughout recombination spanning 820 a.u.–1050 a.u. in the G case and in the U case, respectively. (c) Dependence of harmonic order on ionization time (circles) and emission time (dots) of the electron in the multicycle laser field. The laser parameters are the same as those in Fig. |
4. Conclusions
In this study, we demonstrate a new method to investigate the origin of spectral minimum in HHG. We calculate the HHG spectra generated from the 1D stretched 

Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] | |
| [33] | |
| [34] | |
| [35] | |
| [36] | |
| [37] | |
| [38] | |
| [39] | |
| [40] | |
| [41] | |
| [42] |