


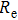
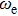
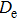


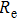
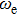
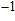
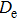

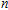
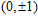




























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Geometries, stabilities, and electronic properties analysis in InnNi(0,±1) clusters: Molecular modeling and DFT calculations
Cite this Article
Shi Shun-Ping, Zhang Chuan-Yu, Zhao Xiao-Feng, Li Xia, Yan Min, Jiang Gang. Geometries, stabilities, and electronic properties analysis in InnNi(0,±1) clusters: Molecular modeling and DFT calculations
. Chinese Physics B, 2017, 26(8): 083103 
Permissions
Geometries, stabilities, and electronic properties analysis in InnNi(0,±1) clusters: Molecular modeling and DFT calculations
† Corresponding author. E-mail:
Project supported by the Cultivating Program of Excellent Innovation Team of Chengdu University of Technology (Grant No. KYTD201704), the Cultivating Program of Middle-aged Backbone Teachers of Chengdu University of Technology (Grant No. 10912-KYGG201512), the National Natural Science Foundation of China (Grant No. 11404042), the Science Fund from the Science & Technology Department of Sichuan Province, China (Grant No. 2016RZ0069), and the Research Foundation of Chengdu University of Technology, China (Grant No. 2017YG04).
Abstract
Density functional theory (DFT) with the B3LYP method and the SDD basis set is selected to investigate In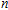
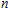

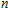
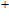
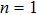
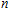
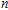
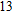
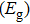
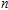
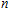
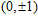
Keyword:
n
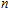
Ni
(
0
,
±
1
)
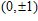
clusters" target=_blank>In
n
Ni
(
0
,
±
1
)
clusters;stability;electronic property;density functional theory
1. Introduction
Atomic clusters play a bridge between molecular and condensed matter physics. Therefore, the properties of clusters are widely studied as a function of size and composition with two objectives: First, researcher hopes to learn how they evolve toward bulk properties and to find unique properties for specific cluster sizes that differ largely from their bulk counterparts. Second, when the composition varies, corresponding to the structure, stability, electronic property, magnetic moment, etc. are also changed. These characteristics make a huge potential of clusters studies for a wide range of applications. In recent years, semiconductor nanomaterials have drawn a lot of research efforts both for theoretical and for their potential technological applications. Indium as a good semiconductor element, the technological importance is growing due to its potential mainstream use in future nanoelectronic devices.[1] Thus, over the past decades, the structural and electronic properties of indium clusters have been extensively studied both experimentally and theoretically.[2–11] For example, Staudt and Wucher[11] investigated the yields of neutral and charged In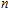
However, a great number of experimental and theoretical works about doped indium clusters have been studied.[12–31] This is motivated by the fact that an atom doped in a small indium cluster can strongly change the properties of the pure indium cluster. Janssens et al.[12] performed the structures and ionization potentials of small In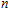


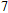

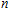


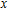
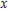
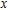
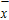
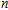

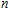
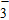


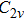


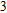
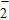
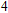

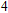

All the time, TM element-doped clusters have attracted attention, because when a transition metal is encapsulated into clusters, some special phenomena appear, for example, when the Ti atom is doped to Ga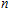
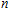

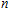
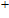
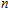
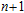
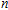
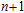
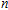
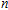
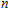
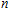

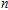
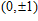


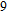
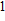
2. Computational details
A research is performed for the lowest energy and low-lying structures of In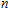
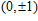
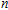
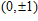


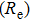
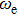
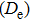
| Table 1.
Calculated values of bond length |
For small-sized neutral, cationic, and anionic In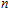
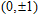
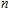
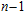
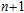
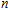
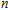
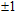
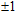


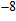
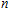
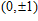
3. Results and discussion
3.1. Geometrical structures of In
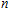
Ni
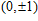
(n = 1–14) clusters
For In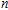
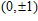
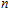
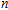
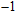

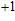
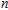
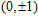
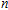
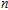
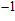
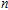
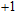
 | Fig. 1. (color online) The lowest energy and low lying structures of In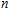
|
 | Fig. 2. (color online) The lowest energy and low lying structures of In

|
 | Fig. 3. (color online) The lowest energy and low lying structures of In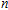
|
(i) InNi, InNi
The equilibrium geometries of InNi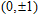
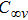
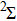


(ii) In



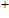
The possible structures such as 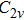
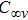
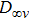

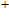

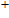

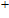
(iii) In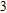
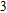

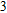
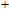
In the case of n = 3, In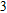
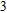

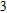


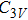
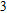
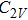
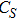
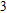
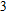
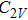
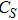
(iv) In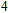
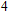

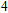
For n = 4, the lowest-energy structures of the neutral, cationic, and anionic In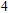

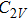
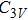

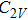


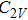
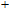
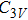
(v) In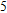
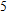

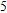
Starting n = 5, the lowest energy structures for neutral and cationic forms are similar, but the most stable structure of anionic cluster is different with neutral and cationic forms, although the symmetries of In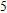
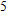

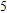
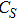


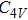
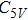
(vi) In



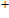
By calculations, we obtain that the spin triplet state is the lowest energy for the In
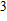
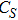



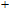

(vii) In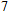
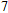

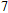
The global minimum of In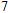

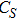

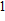


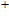


(viii) In



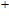
In the case of the most stable In
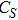




(ix) In



The ground state structure obtained for In
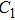





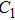


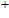

(x) In
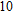

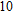
The lowest energy structures of neutral, cationic, and anionic In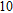
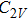

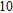
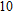
(xi) In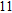
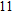


Starting from various initial structures, the In

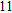


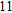
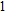
(xii) In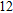


When the size of In


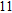

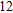

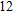
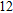
(xiii) In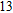
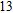

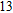
The lowest energy structure for neutral In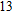



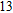
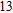
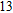

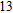
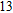
(xiv) In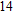
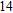

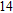
Finally, the ground state structure of In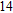
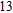
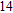



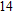
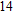

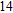
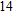
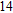

3.2. Relative stabilities
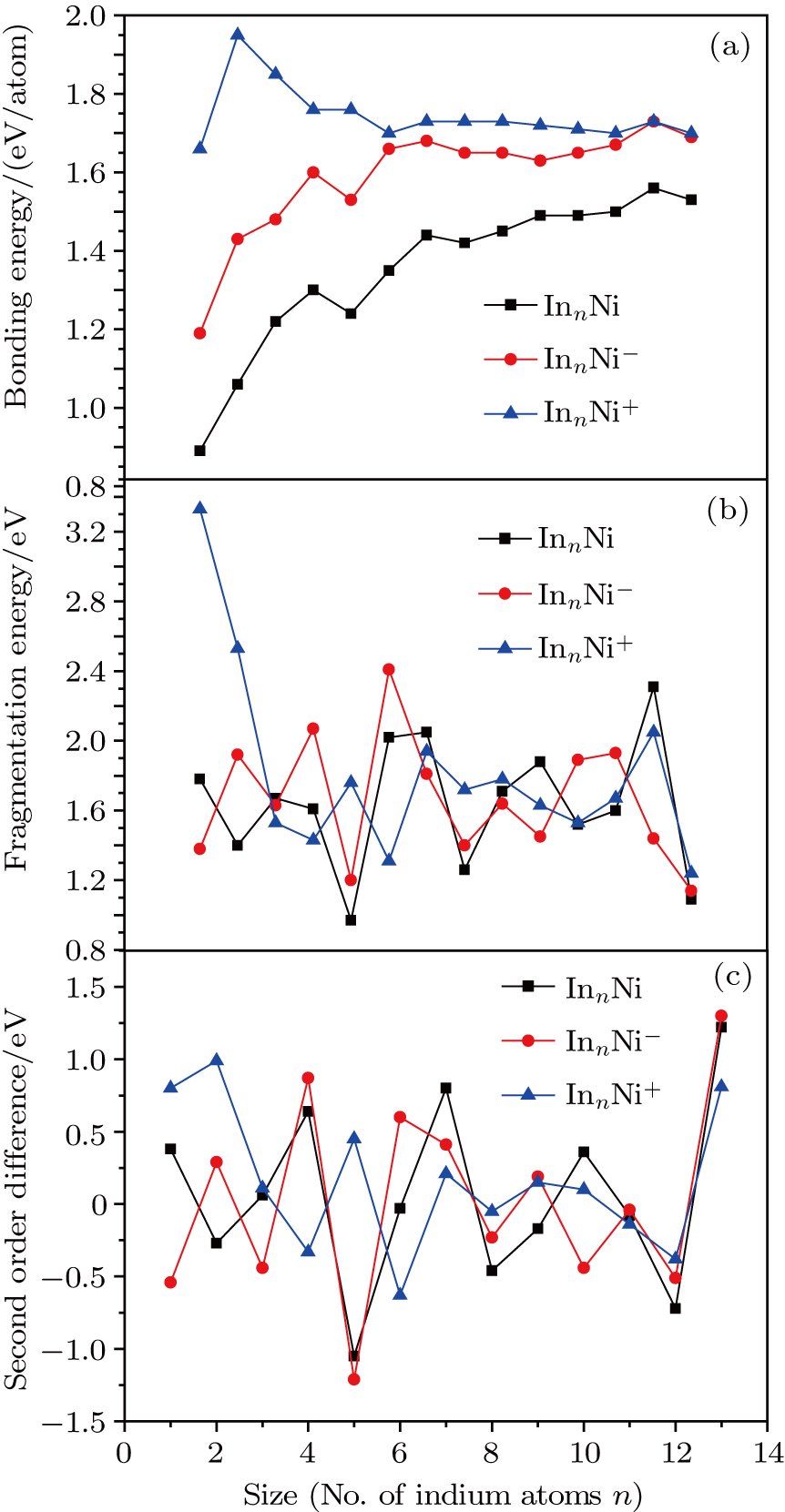
In order to investigate the strength of chemical bonds, the relative abundances determined in mass spectroscopy experiments, and the thermodynamic stabilities of most stable In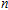
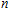

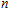
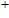
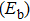
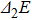
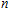
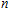

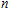
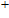
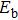
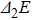
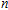
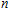


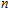
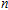

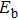
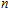
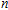

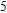
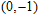

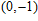

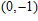
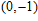

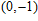
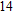
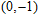
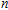

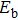
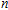
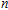
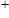
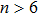
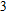

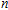
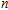

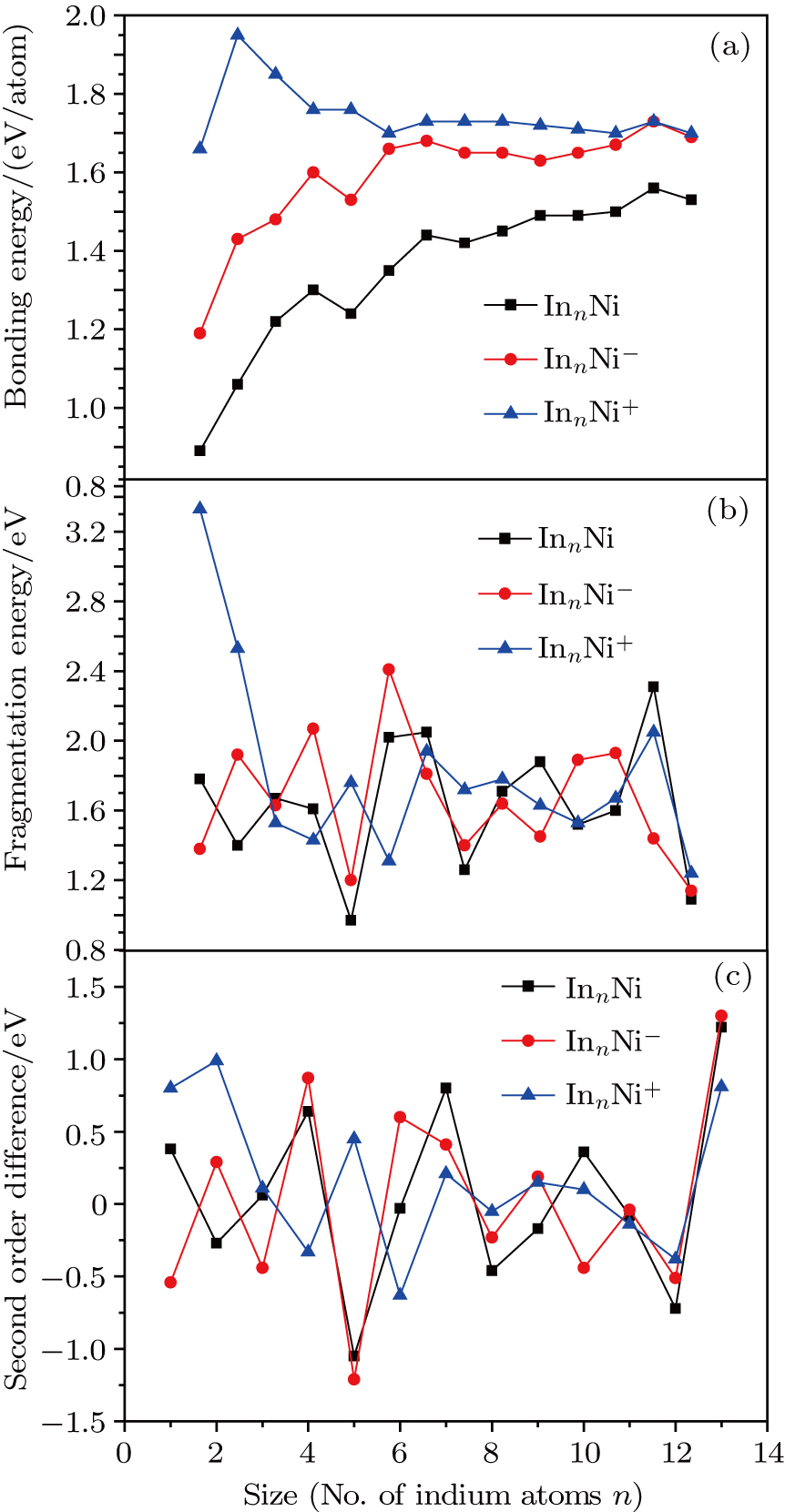 | Fig. 4. (color online) Binding energies, fragmentation energies, and the second-order energy difference for the In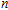
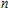

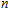
|
As shown in Fig. 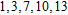
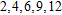





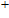
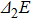
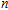
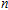

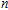
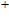
3.3. Electronic properties
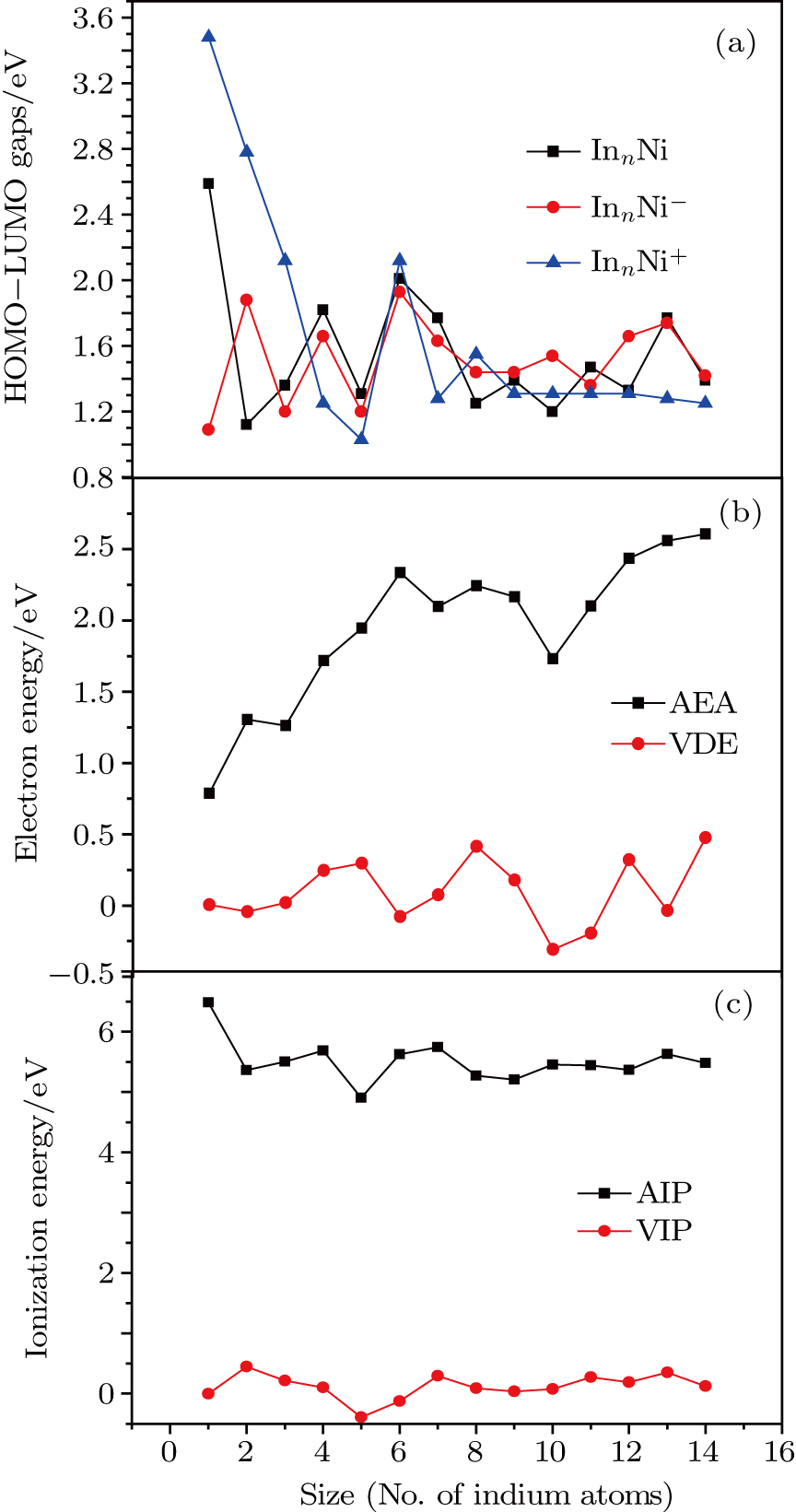
To obtain the typical electronic properties of In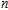
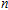

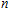
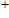
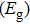
As presented in Fig. 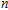
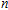

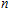
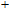
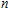
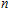
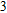
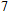




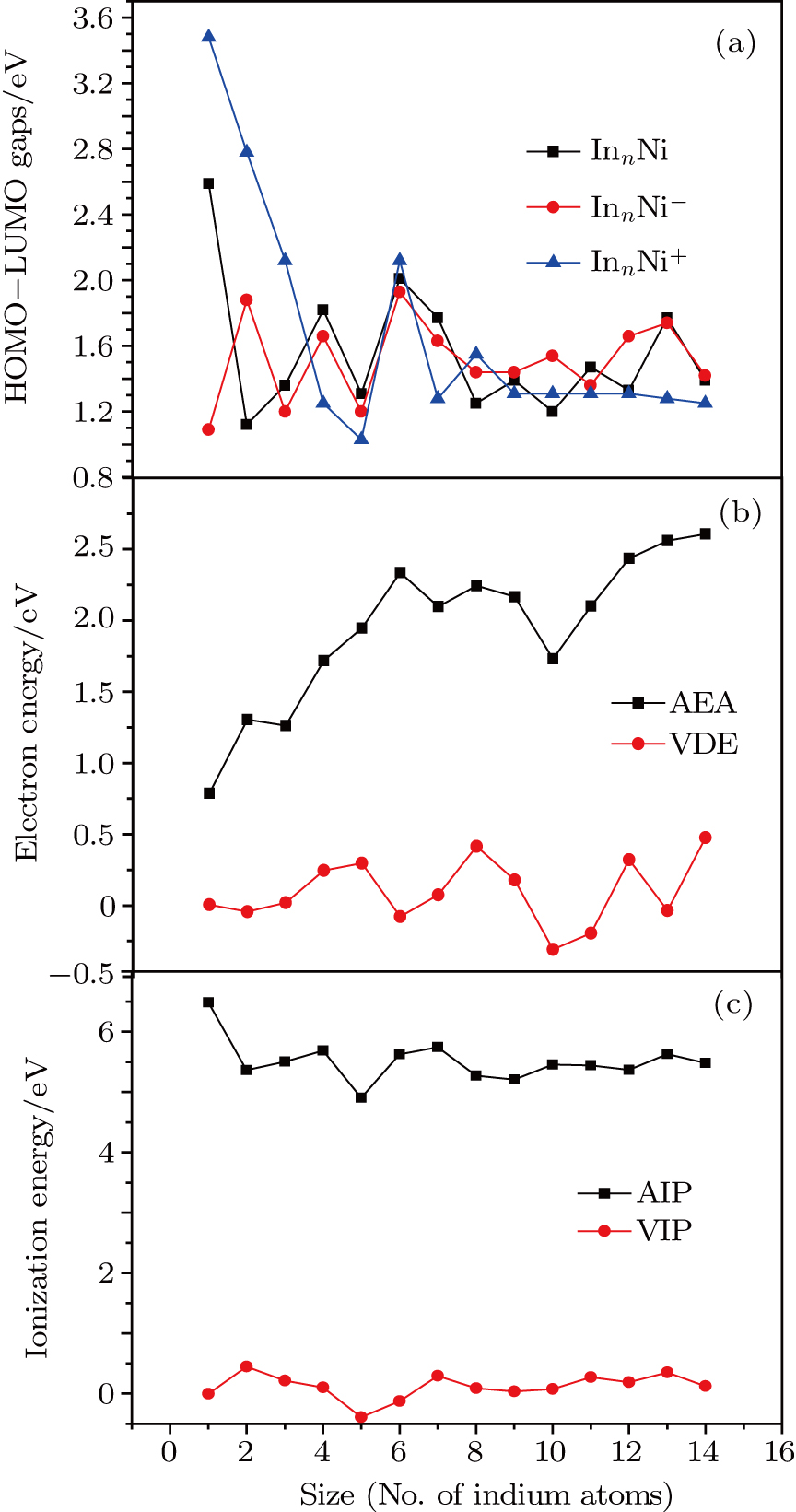 | Fig. 5. (color online) HOMO–LUMO gaps, electron energies, and ionization energies for the In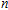
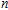

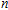
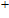
|
Other important electronic properties that reflect the stability of clusters are their electron affinities and ionization potentials.[50] Comparing the total energies of In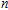
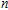

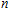
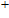
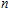
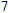

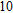






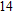

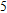

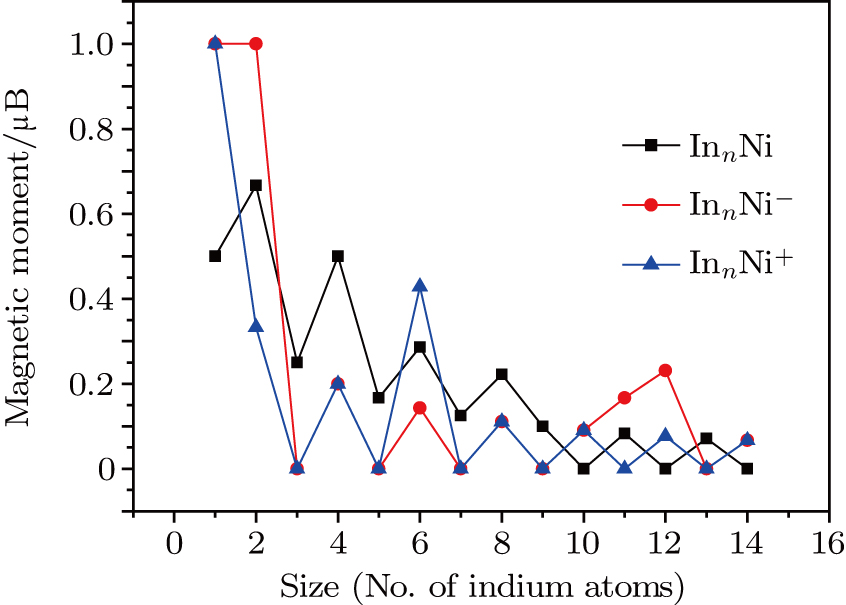
3.4. Magnetisms
The total magnetic moments of the studied In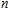
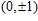
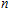

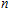
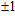
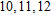


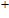
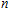
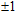
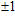


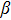
 | Fig. 6. (color online) Magnetic moment for the In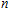
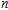

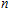
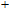
|
3.5. Natural population analysis
The localization of charge and the charge-transfer information of In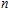
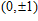
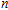
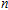
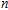
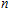
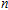
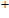
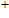

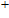
| Table 2.
The total charges (Q) of In atoms and the charges (Q) of Ni atom for the ground state structures of In |
4. Conclusions
In this work, we performed a systematic study of the neutral, anionic, and cationic In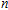
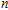
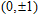
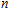

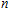
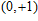




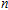


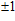
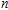
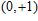
Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] | |
| [33] | |
| [34] | |
| [35] | |
| [36] | |
| [37] | |
| [38] | |
| [39] | |
| [40] | |
| [41] | |
| [42] | |
| [43] | |
| [44] | |
| [45] | |
| [46] | |
| [47] | |
| [48] | |
| [49] | |
| [50] | |
| [51] | |
| [52] | |
| [53] | |
| [54] |