



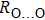
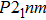
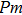
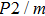
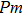
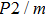


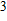
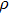


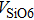
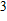
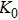
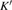
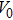
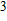


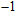
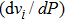
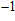
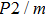
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Structural, elastic, and vibrational properties of phase H: A first-principles simulation
Cite this Article
Lv Chao-Jia, Liu Lei, Gao Yang, Liu Hong, Yi Li, Zhuang Chun-Qiang, Li Ying, Du Jian-Guo. Structural, elastic, and vibrational properties of phase H: A first-principles simulation. Chinese Physics B, 2017, 26(6): 067401 
Permissions
Structural, elastic, and vibrational properties of phase H: A first-principles simulation
† Corresponding author. E-mail:
Abstract
Phase H (MgSiO
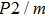
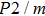
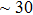
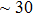

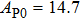
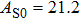

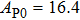
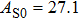
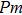
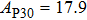
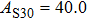
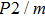
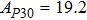
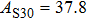
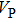
1. Introduction
The presence of water in the lower mantle has various functions, such as dramatically lowering the melting temperature of mantle rocks, causing arc magmatism, reducing the magma viscosity and density, enhancing magma migration, influencing the elastic and rheological properties of the mantle, and broadening seismic discontinuities.[1] It is now accepted that water can percolate through the Earth’s interior by subducting slabs through hydrous minerals, such as serpentine.[2,3] The serpentine can transform into several dense hydrous magnesium silicates (DHMSs), depending on the pressure and temperature conditions.[4–6] Since the first discovery of 10-Å phase,[7] dense hydrous magnesium silicates including 3.65-Å phase, phase A, phase B, superhydrous phase B, phase D, phase E, etc. have received considerable attention because of their presumed role in the water resources of the Earth.[8–10]
Phase D (MgSi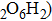


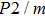
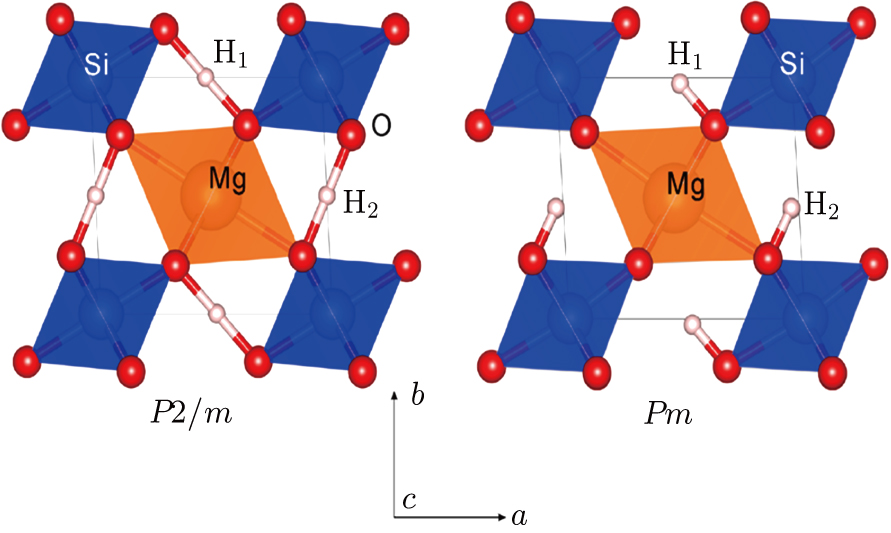
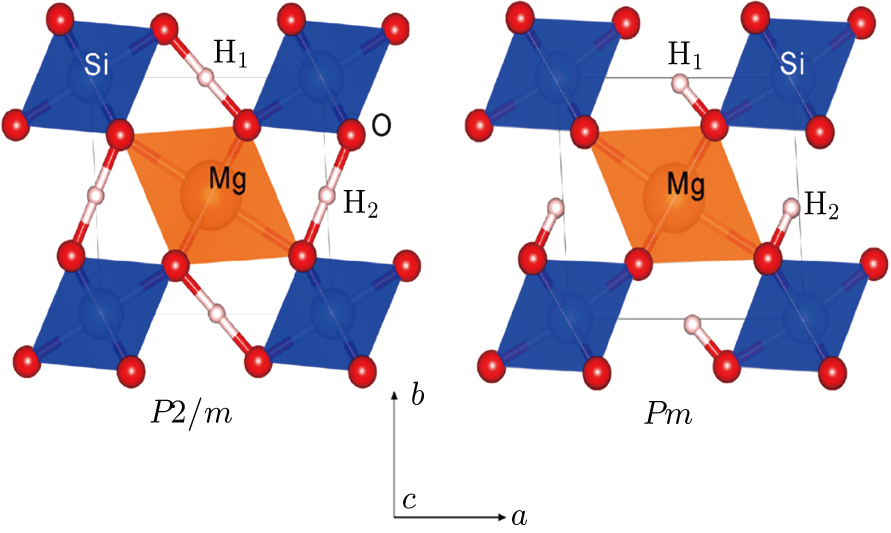 | Fig. 1. (color online) Structures of phase H with 
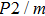
|
In this study, in order to further understand the stability of phase H under high pressure, the crystal structure, elastic constants, seismic velocities and anisotropies of the two different possible symmetry phase H’s (Pm and 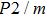

2. Methods
2.1. Computational methods
The first-principles calculations were performed using density functional perturbation theory (DFPT),[18] density functional theory (DFT),[19,20] and the plane wave pseudopotential method, as implemented in the CASTEP codes.[21] The generalized gradient approximation (GGA) with PBE parameterization was adopted to describe the exchange-correlation functional.[22] GGA functional was tested to provide a better performance than local density approximation (LDA) for simulations of hydrogen bonding.[23,24] Norm-conserving pseudopotentials[25] were employed to model the electron–ion interaction. The electronic wave functions were expanded in plane-waves with a kinetic energy cutoff of 1200 eV. The irreducible Brillouin zone of the phase H is sampled on a 5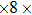
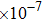
The structures of phase H used in this study possess monoclinic structures with 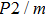
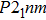
2.2. Benchmark calculations
To assess the performances of the DFT and DFPT total-energy approach in our study, test calculations of the cell parameters and elastic constants of phase H were performed. The differences in cell volumes, lengths of a, b, and c axes between the calculated results and corresponding experimental values[16] are 0.1%, 1.2%, 0.7%, and 0.4% respectively, or 2.7%, 1.7%, 0.6%, and 0.7% compared with the other experimental results[15] (Table 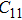
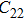
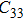
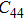
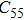
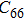

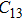
| Table 1.
Benchmark calculations results. . |
3. Results
3.1. Crystal structure
Ever since the first-principles prediction of this new mineral was proposed,[14] phase H has been observed in the two separate experimental research studies.[15,16] However, due to the difficulties in detecting the positions of hydrogen atoms and pressure-induced rapid amorphization of the sample,[15,16] the structure of phase H was roughly judged to be an orthorhombic system with symmetry 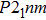
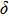

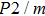

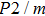
Geometry optimization has been conducted on both Pm and 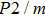


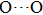
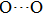
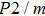
| Table 2.
Structural properties of phase H. . |
 | Fig. 2. (color online) Lattice parameters of phase H each as a function of pressure: (a) 
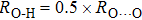
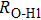
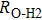
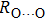
|
The volume–pressure relationships (Fig. 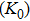
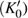
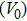
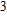
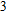
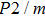
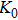
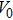
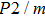
3.2. Elasticity
where
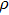
is the mass density determined by the equilibrium of crystal lattice from the first-principles optimization, K is the bulk modulus, and G is the shear modulus. The
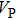
and
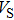
of phase H with
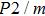
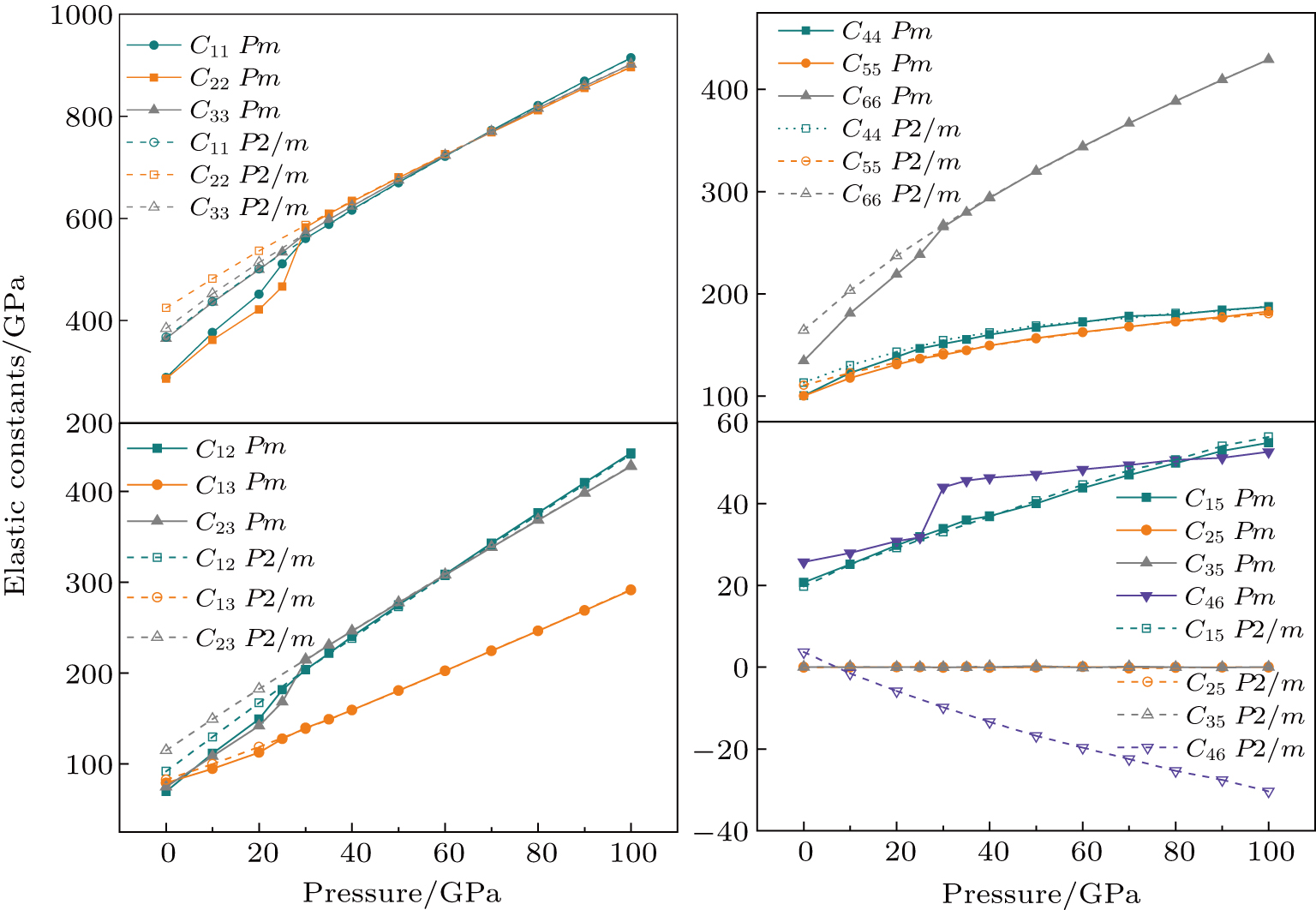
symmetry increase smoothly with increasing pressure. The
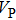
and
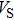
of Pm symmetry sharply increase from 25 GPa to 30 GPa (Fig. 4 ) compared with those of
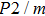
. The pressure dependent
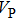
and
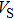
of phase H with Pm and
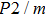
symmetry show similar tendencies to those of K and G. At pressures above 30 GPa, the values of
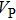
and
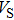
of Pm and
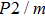
symmetry are almost identical. The symmetrization of hydrogen bonds also generally takes place in plenty of minerals under high pressures, such as ice,
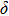
-AlOOH and hydrous silica.[36,38] Therefore, the symmetrization of hydrogen bonds in dense hydrous silicate may contribute to the abnormally pressure-dependent
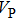
and
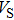
in the upper mantle. The
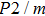
symmetry shows higher density and velocity than the Pm symmetry under pressure lower than 30 GPa. The transition from Pm to
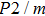
may somewhat contribute to the leaps of density and seismic wave velocity in the transition layer of the mantle.
The pressure-dependent elastic constants for the two phase H are shown in Fig. 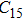
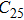
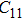
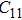
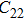


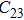
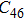
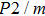
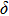
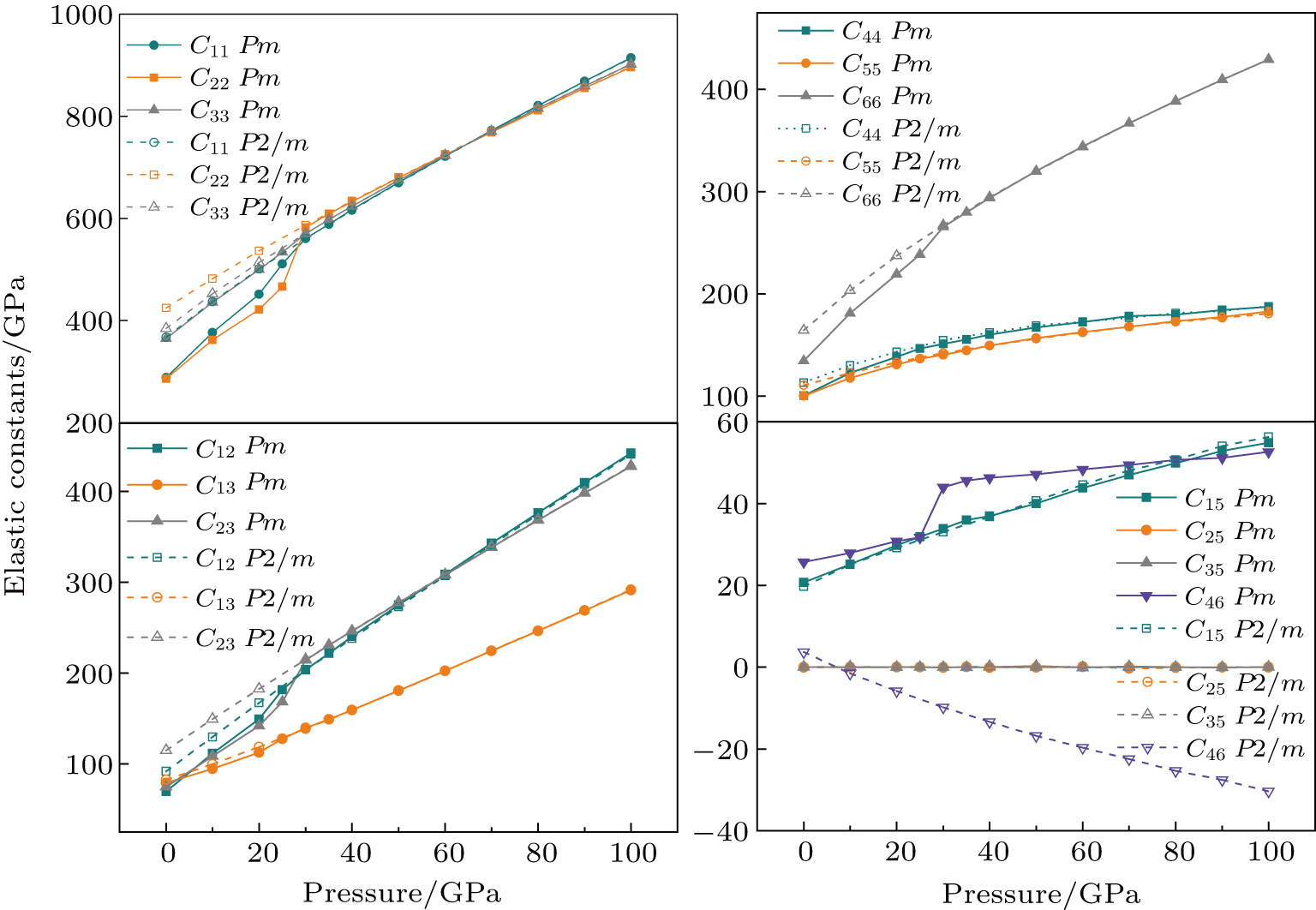 | Fig. 3. (color online) Pressure-dependent elastic constants for phase H. |
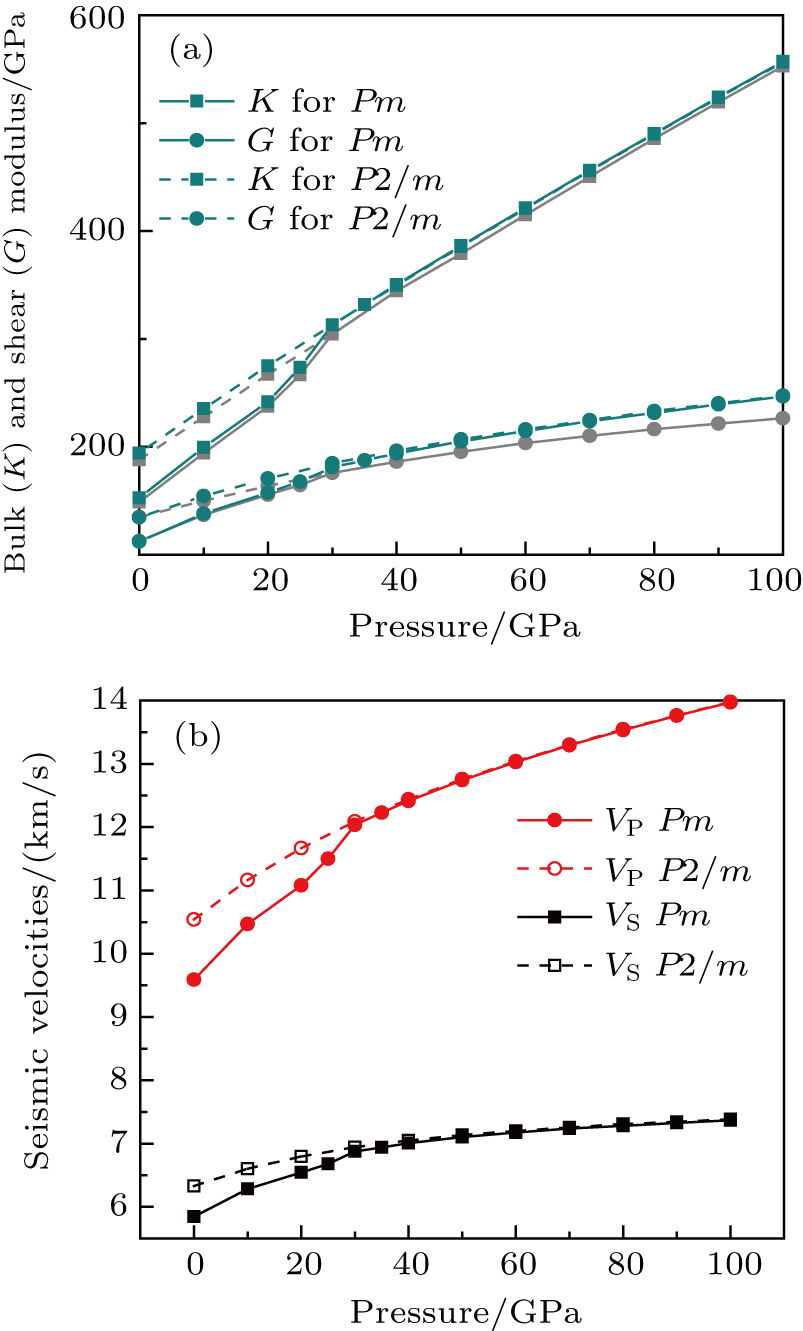
Bulk (K) and shear (G) moduli are determined by the Voigt–Reuss–Hill average (Fig. 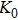
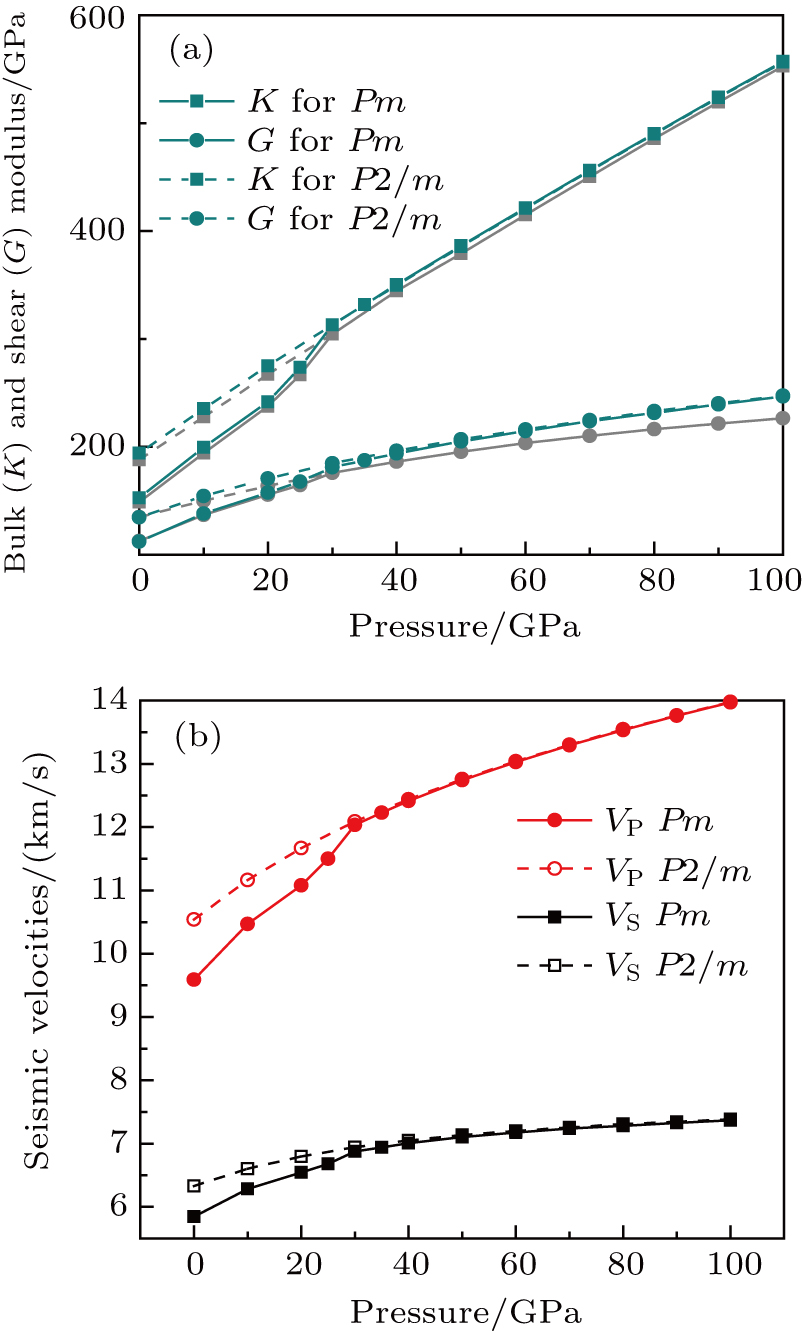 | Fig. 4. (color online) Bulk and shear moduli (a) and seismic velocities (b) of phase H as a function of pressure. Grey lines and points in panel (a) stand for K and G from Ref. [17]. |
Sound wave velocities from seismic observation can reflect the composition and structure of the mantle. For crystalline material, the compressional wave velocity 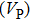
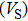
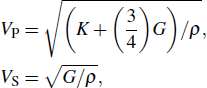 |
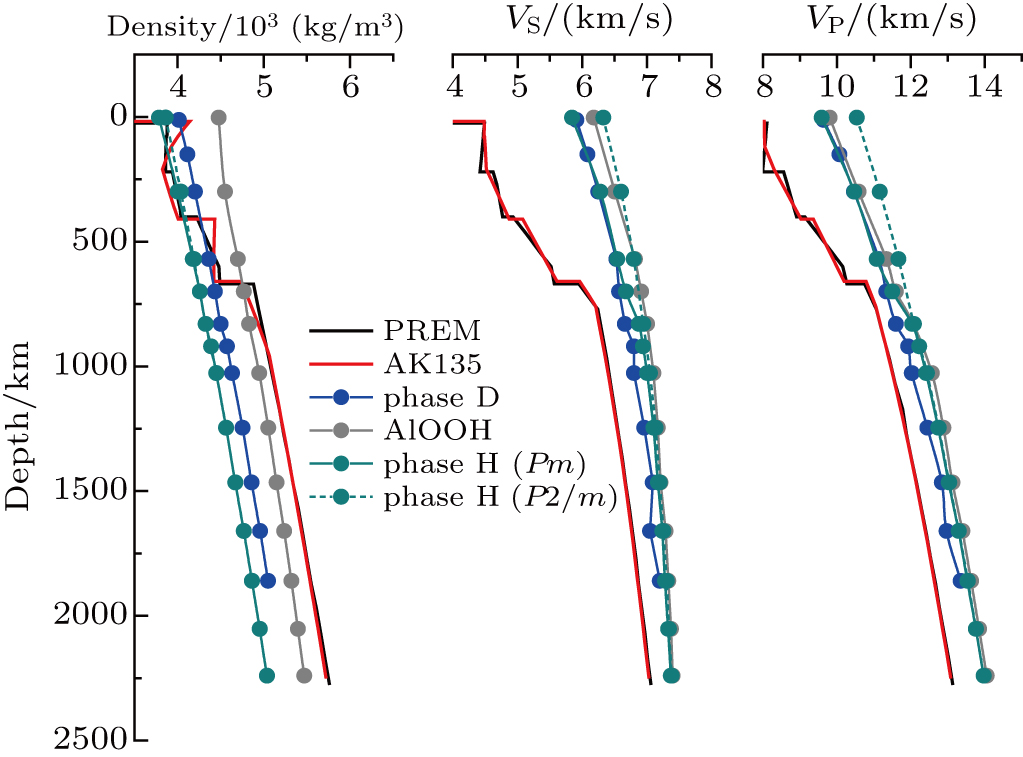
The values of density, 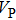

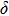
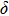
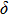
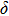
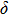

 | Fig. 5. (color online) Densities and seismic wave velocities of phase H, phase D and 
|
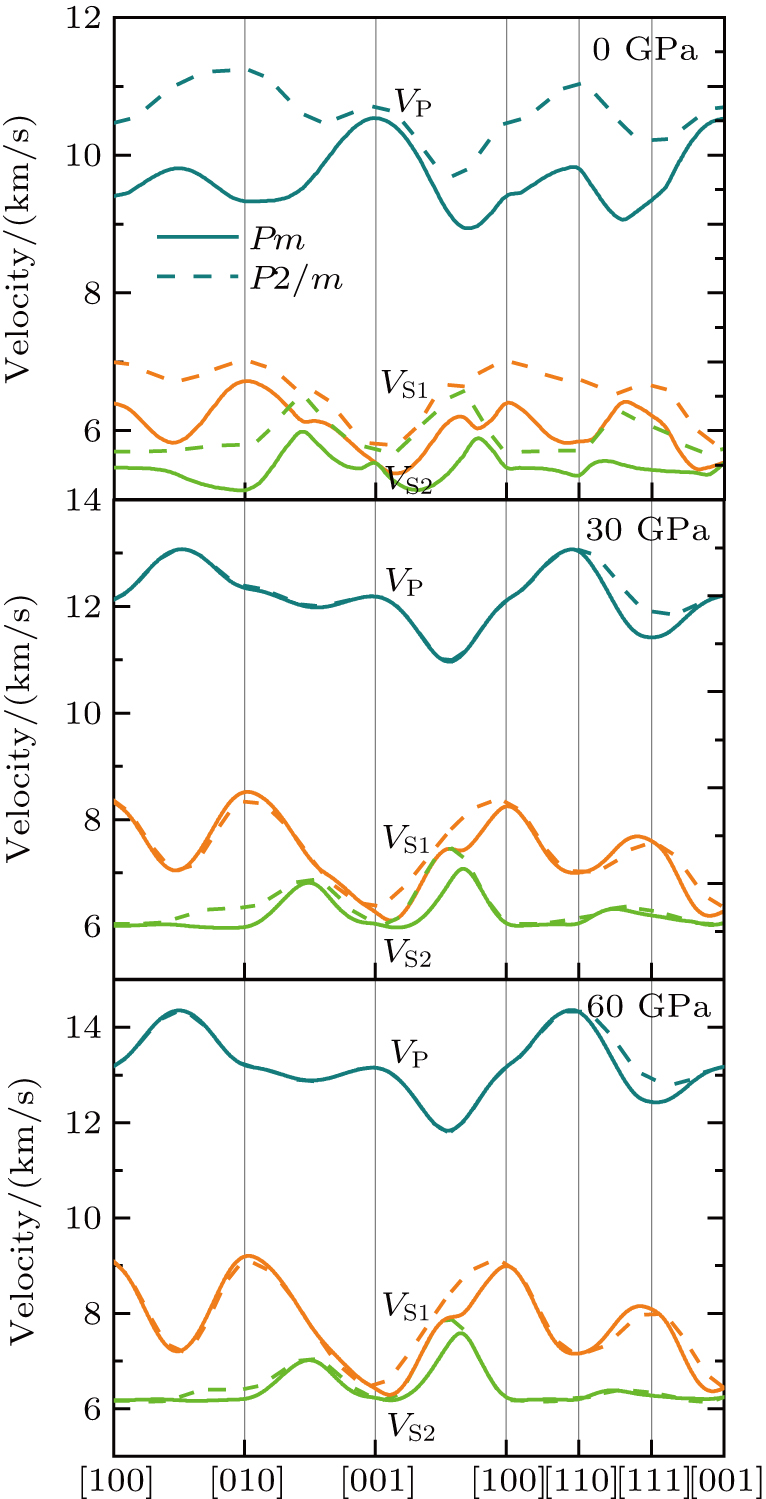
Single crystal seismic velocities for phase H are also calculated by solving Cristoffel’s equation:[42]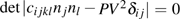
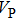
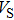
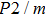
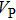
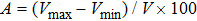
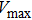
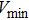

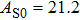
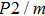
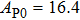
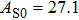
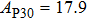
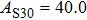
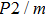
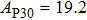
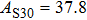
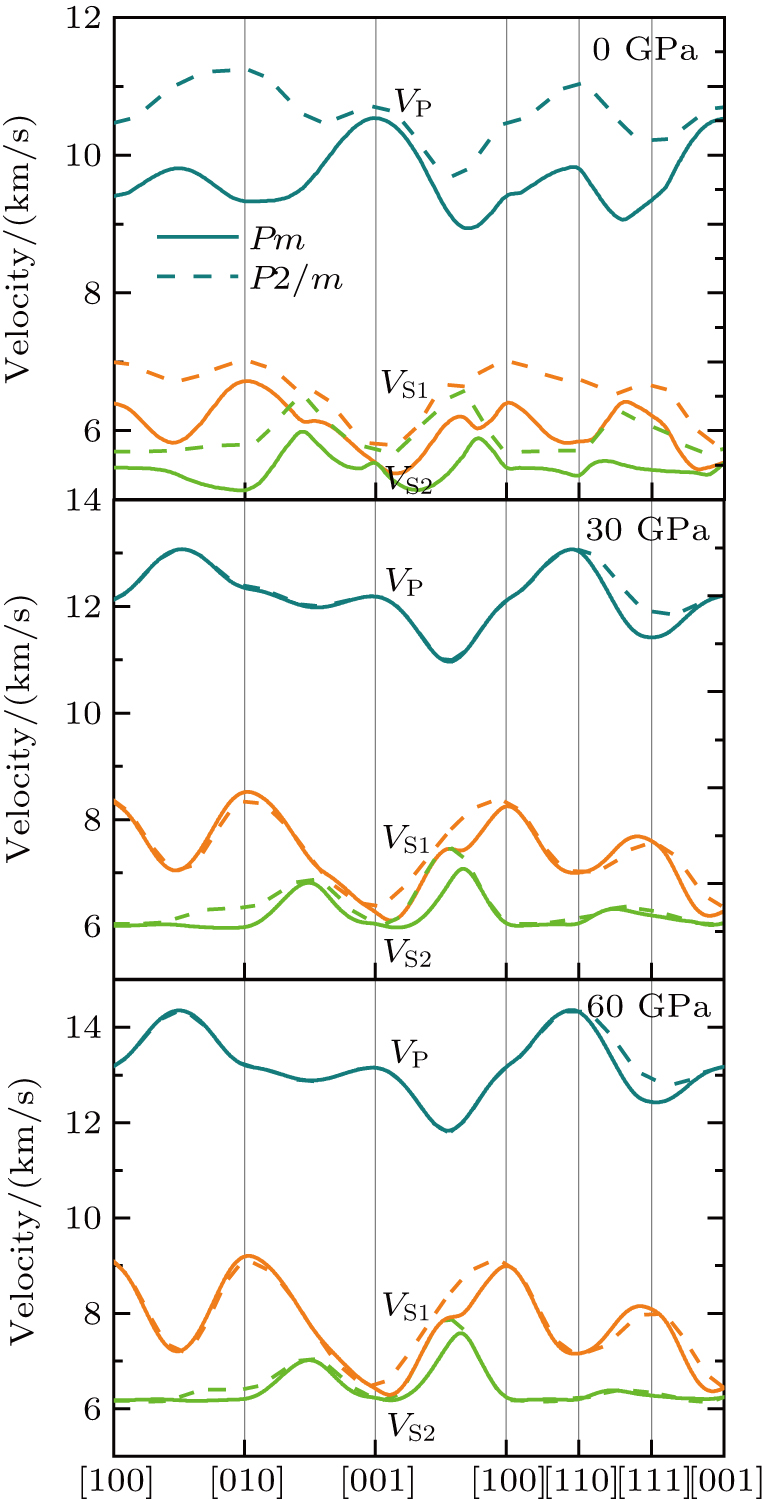 | Fig. 6. (color online) Single crystal seismic velocities of phase H under 0, 30, and 60 GPa. |
3.3. Vibration
Vibrational spectra of crystalline solids can be interpreted by using the factor group analysis.[44] For phase H with 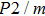
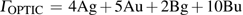
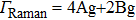
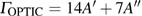
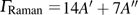
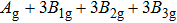
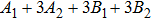

Raman frequencies of the two phase H structures are calculated and plotted each as a function of pressure in Fig. 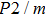
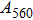
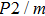
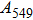
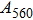
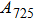
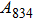
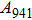
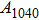

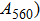
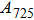
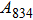

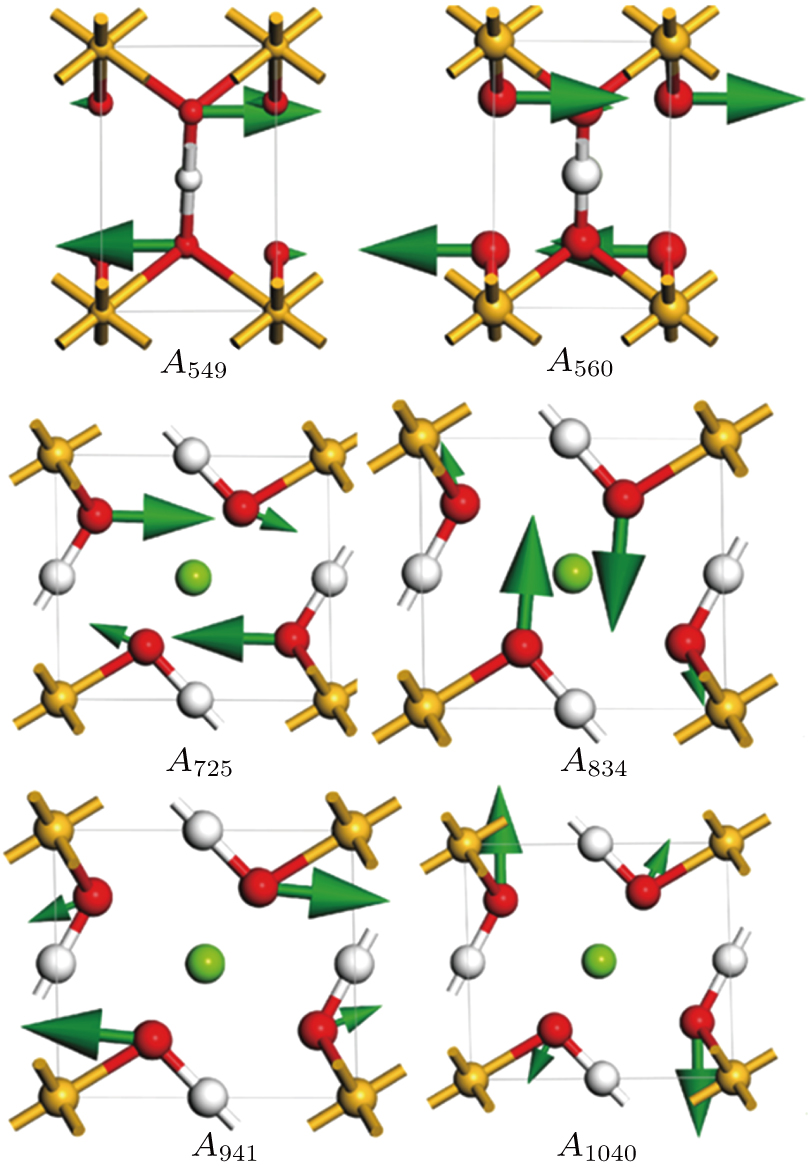
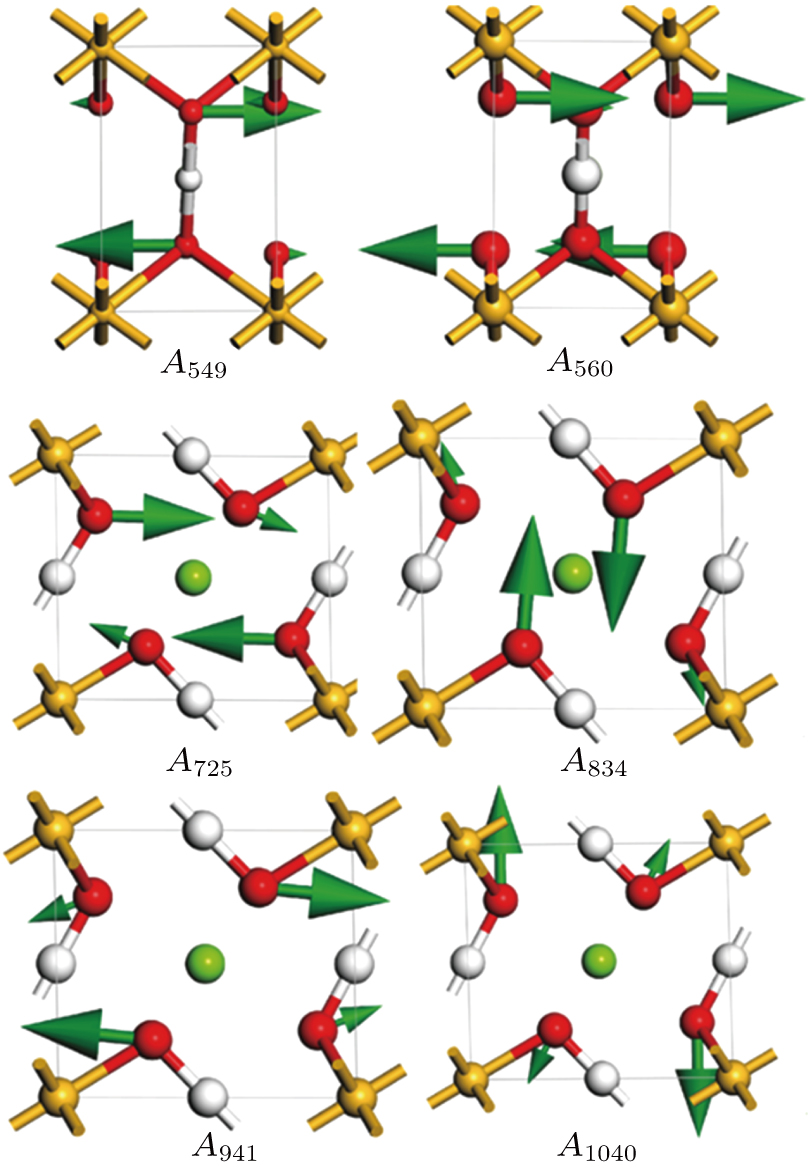 | Fig. 7. (color online) Vibrational patterns of the 
|
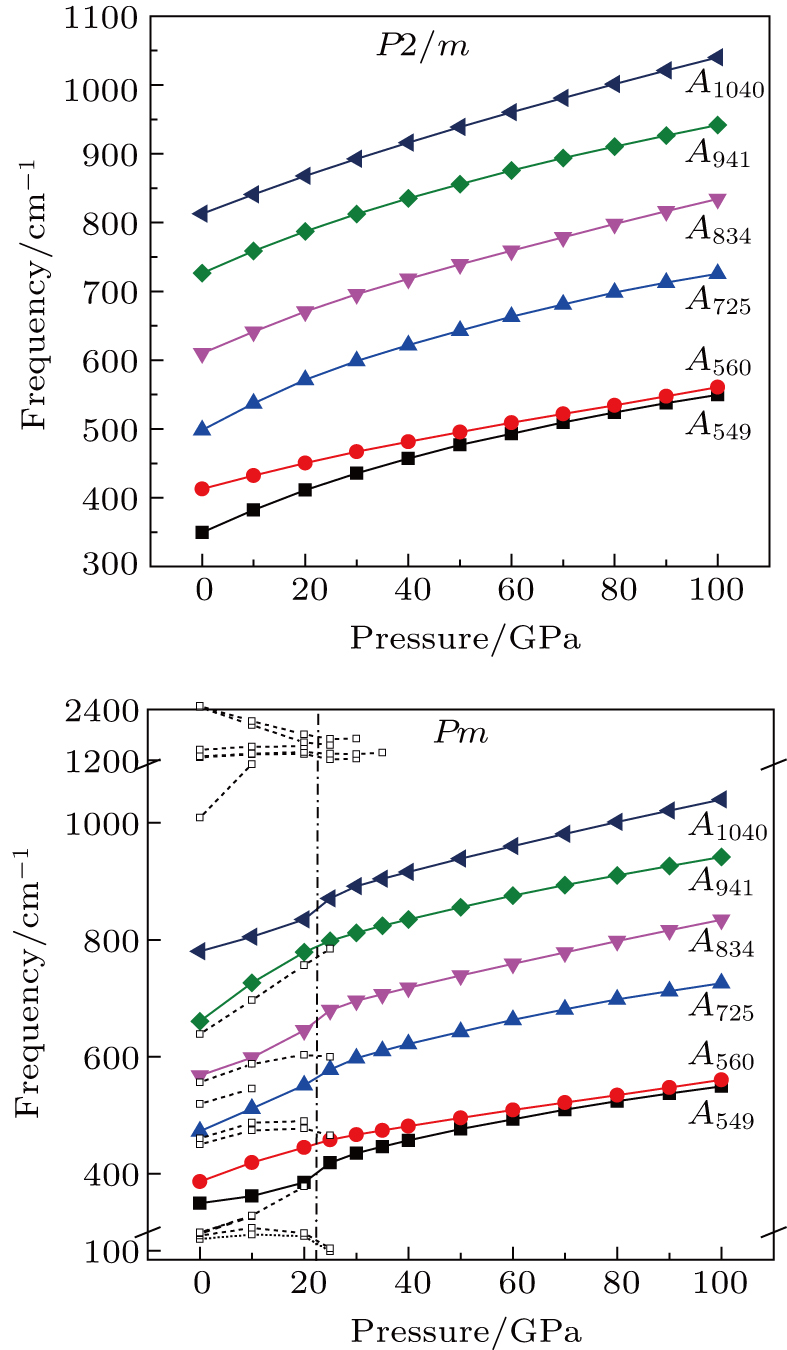
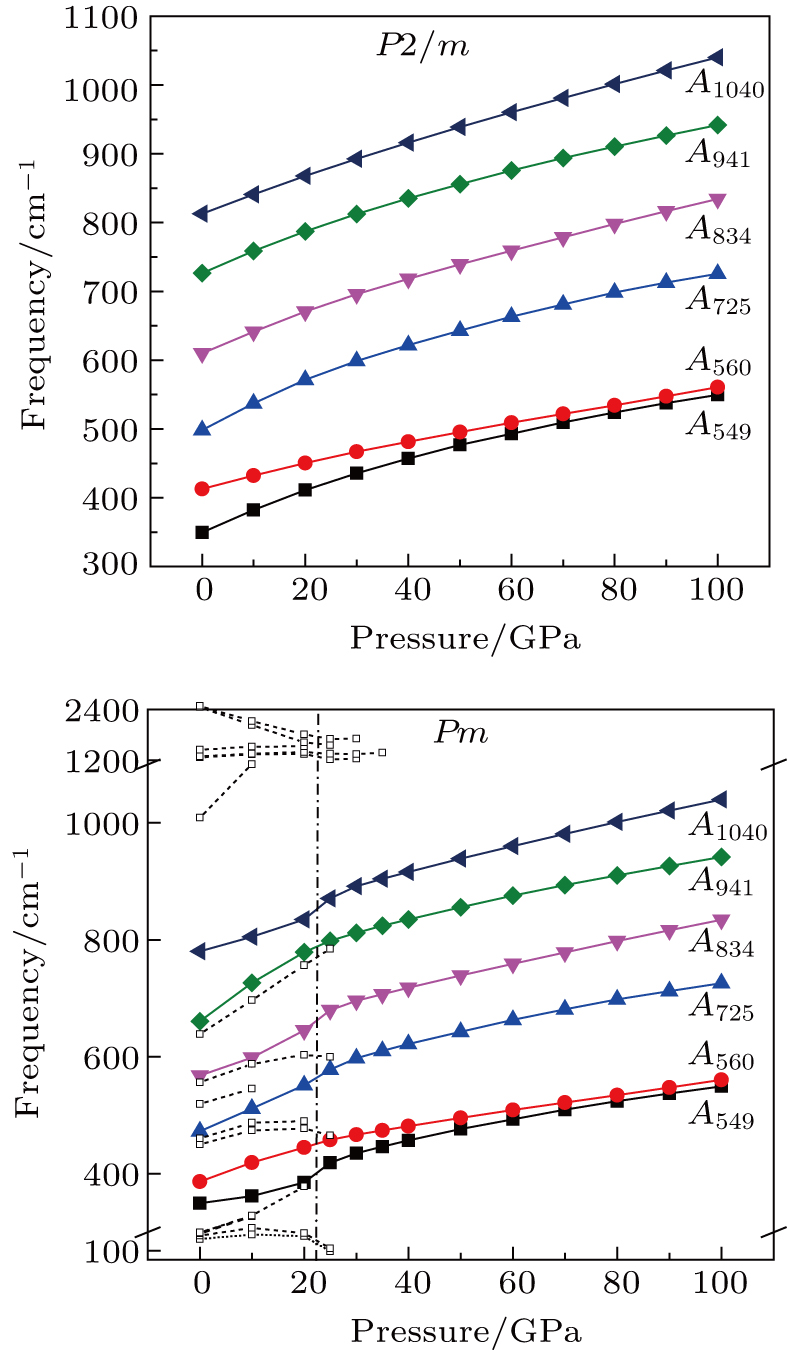 | Fig. 8. (color online) Plots of Raman-active frequency of phase H versus pressure. Dasheed plots and lines refer to the Raman modes which are inactive under high pressures. |
At 0 GPa, 21 Raman modes are observed in phase H with Pm symmetry. These modes can be classified as two groups. Group 1 includes 15 vibrational modes with frequencies ranging from 226 cm
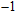
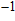
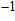
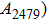
The vibrational patterns of Pm symmetry can be divided into two stages: from 0 GPa to 30 GPa and from 30 GPa to 100 GPa (Fig. 
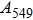
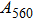
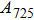
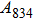
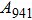
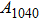
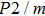

Six Raman modes: 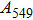
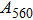
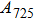
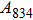
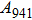

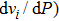
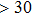
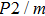
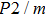
| Table 3.
Pressure shifts of the Raman modes of phase H. . |
4. Conclusions and perspectives
The phase H (MgSiO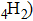
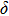
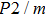
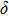
The 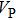
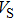
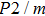
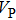
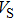
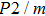
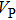
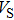
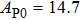
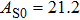
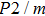



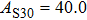
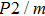
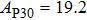
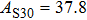
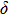
Raman spectra of phase H show the effects of symmetrization of hydrogen bonds on vibrational frequency and intensity. At 0 GPa, 21 Raman modes are observed in phase H with Pm symmetry. As for 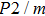
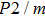
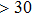
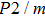
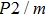
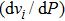
Reference
| [1] | |
| [2] | |
| [3] | |
| [4] | |
| [5] | |
| [6] | |
| [7] | |
| [8] | |
| [9] | |
| [10] | |
| [11] | |
| [12] | |
| [13] | |
| [14] | |
| [15] | |
| [16] | |
| [17] | |
| [18] | |
| [19] | |
| [20] | |
| [21] | |
| [22] | |
| [23] | |
| [24] | |
| [25] | |
| [26] | |
| [27] | |
| [28] | |
| [29] | |
| [30] | |
| [31] | |
| [32] | |
| [33] | |
| [34] | |
| [35] | |
| [36] | |
| [37] | |
| [38] | |
| [39] | |
| [40] | |
| [41] | |
| [42] | |
| [43] | |
| [44] | |
| [45] | |
| [46] | |
| [47] | |
| [48] | |
| [49] | |
| [50] |