Liu Ya-Hui, Chong Xiao-Yu, Jiang Ye-Hua, Feng Jing. Stability, electronic structures, and mechanical properties of Fe–Mn–Al system from first-principles calculations. Chinese Physics B, 2017, 26(3): 037102
Permissions
Stability, electronic structures, and mechanical properties of Fe–Mn–Al system from first-principles calculations
Liu Ya-Hui, Chong Xiao-Yu, Jiang Ye-Hua †, Feng Jing
Faculty of Materials Science and Engineering, Kunming University of Science and Technology, Kunming 650093, China
The stability, electronic structures, and mechanical properties of the Fe–Mn–Al system were determined by first-principles calculations. The formation enthalpy and cohesive energy of these Fe–Mn–Al alloys are negative and show that the alloys are thermodynamically stable. Fe3Al, with the lowest formation enthalpy, is the most stable compound in the Fe–Mn–Al system. The partial density of states, total density of states, and electron density distribution maps of the Fe– Mn–Al alloys were analyzed. The bonding characteristics of these Fe–Mn–Al alloys are mainly combinations of covalent bonding and metallic bonds. The stress-strain method and Voigt–Reuss–Hill approximation were used to calculate the elastic constants and moduli, respectively. Fe2.5Mn0.5Al has the highest bulk modulus, 234.5 GPa. Fe1.5Mn1.5Al has the highest shear modulus and Young’s modulus, with values of 98.8 GPa and 259.2 GPa, respectively. These Fe–Mn–Al alloys display disparate anisotropies due to the calculated different shape of the three-dimensional curved surface of the Young’s modulus and anisotropic index. Moreover, the anisotropic sound velocities and Debye temperatures of these Fe–Mn–Al alloys were explored.
Iron, manganese, and aluminum are the main components of low density, high strength steel. The Fe–Mn–Al alloys have recently attracted much attention because these alloys have excellent corrosion resistance, outstanding mechanical properties, and low density.[1,2] In addition, the Fe–Mn–Al system is known as an effective substitute for stainless steel at high temperature oxidation resistance.[3] The Fe–Mn–Al alloys also provide excellent mechanical properties and can serve as heat-resistant steel, cryogenic steel, magnetic material, and nonmagnetic stainless steel.[4] Earlier research shows the Fm3m type DO3 [(Fe,Mn)3Al] phases distributing in ferrite in the Fe–Mn–Al–C steel; the ordered phases have a reinforcement effect in the Fe–Mn–Al–C steel.[5] Therefore, we here investigate the stability, electronic structures, and mechanical properties of the Fe–Mn–Al system. Some Fe–Mn–Al alloys have been investigated by experiments and theory in recent years. Yang et al.[6] studied the tensile deformation of low density Fe–Mn–Al–C steel. Zhang et al.[7] investigated the elastic properties of ferromagnetic body-centered cubic Fe–Mn–Al alloys. Lindahl et al.[8] studied the ordering in ternary BBC alloys applied to the Al–Fe–Mn system. Umino et al.[9] studied the phase equilibria in the Fe–Mn–Al-system by the experimental determination and thermodynamics. Yan et al.[10] investigated the anomalous behavior of electrical resistivity in Fe–Mn–Al alloys. Rico et al.[11] studied the magnetic and structural properties of mechanically alloyed Fe0.70−xAl0.30 (x = 0.40 and 0.45) alloys. The above studies focus mostly on phase equilibria, electrical resistivity, and magnetic properties. However, the electronic structures and mechanical properties of the Fe–Mn–Al system still have not been studied systemically. In our research, we employ first-principles calculations based on density functional theory (DFT), investigating the stability, electronic structures, and Debye temperatures of Fe– Mn–Al alloys. Our aim is to obtain complete data of the mechanical properties and electronic structures of the Fe–Mn–Al system. This work will contribute greatly to designing promising high-temperature structural materials, functional materials, composite materials, and high strength materials. The calculated results will help us design different performance materials, and provide theoretical data to design new steel material for us, and so on. The properties of Mn dissolved into Fe3Al ordered phase have no related data in experiments. This work will have a certain reference value.
2. Calculation methods
The crystal structures of the FexMn3−xAl system are shown in Fig. 1. The Fe3−xMnxAl system has a cubic supercell structure with the space group Fm-3m. All of the crystal structures use Mn atoms to replace Fe atoms based on the crystal structure of Fe3Al. All of the variations studied in this paper were computed by first-principles calculations based on density functional theory (DFT), implemented in the Cambridge sequential total energy package (CASTEP) code.[12] Many DFT-based first-principles studies using plane wave ultrasoft pseudopotentials have also been performed in the early studies for different materials.[13,14] The interactions between valence electrons and ionic core were described by ultrasoft pseudopotentials. The exchange correlation energy was treated with generalized gradient approximation (GGA) of the Perdew–Burke–Ernzerhof (PBE) approach.[15] For Fe, Mn, and Al atoms, the electronic states Fe: 3d64s2, Mn: 3d54s2, and Al: 3s23p1 were treated as valence states. The special k-point method proposed by Monkhorst–Pack was used to characterize energy integration in the first irreducible Brillouin zone,[16] and a Monkhorst–Pack k point mesh of 8×8×8 was used for all Fe3−xMnxAl systems. In our calculations, the maximum plane-wave cutoff energy was 450 eV. During the optimization process, the total energy changes finally converged to 2 × 10−6 eV and the forces acting on each atom were reduced to 0.05 eV/Å.
Fig. 1. (color online) Models of Fe3−xMnxAl system (x = 0.5n, n = 0, 1, 2, 3, 4, 5, 6, and x represents the content of Mn). The pink balls represent Al atoms, the green balls represent Mn atoms, and the gray balls represent Fe atoms.
In the CASTEP code, the elastic constants were calculated by the efficient stress–strain method based on Hooke’s law. Several different strain patterns were used to calculate the crystal structure, and the Cauchy stress tensor for each strain mode was evaluated. For a cubic crystal, the related elastic constants can be expressed as[17]
(1)
where σi is the normal stress, τi is the shear stress, Cij is the elastic constant, εi is the shearing strain, and γi is the corresponding normal. The symmetry of the crystal determines the total number of independent elastic constants.
3. Results and discussion
3.1. Stability
The thermodynamic stability of the Fe3−xMnxAl system was evaluated by calculating the formation enthalpy and cohesive energy. In this work, in order to investigate the thermodynamic stability of the FexMn3−xAl system, the two energy formulas can be defined as
(2)
(3)
where ΔHr(FexMnyAlz) and Ecoh(FexMnyAlz) are the formation enthalpy and cohesive energy, respectively; Etot(FexMnyAlz) is the total energy; Eiso(X) is the total energy of a single X atom; and Ebin(X) is the cohesive energy of an X atom in the bulk state.
In this process, we can obtain the lattice parameters after optimizing the crystal structure. The lattice parameters, formation enthalpy, and cohesive energy of Fe, Mn, Al, and the FexMn3−xAl system are listed in Table 1. Obviously, the calculated results show good agreement with other experimental and calculated values.[18–28] The slight deviations may result from the different calculation methods or experimental conditions. The stability of these FexMn3−xAl compounds can be estimate by the formation enthalpy and cohesive energy. In general, the lower the formation enthalpy, the better the thermodynamic stability of compounds. As can be seen from Table 1, all calculated formation enthalpy and cohesive energy are negative, which shows that these FexMn3−xAl alloys are thermal stable.
Table 1.
Table 1.
Table 1.
The calculated lattice parameters (Å), cohesive energy Ecoh (eV/atom), and formation enthalpy ΔHr (eV/atom) of the Fe–Mn–Al system.
The calculated lattice parameters (Å), cohesive energy Ecoh (eV/atom), and formation enthalpy ΔHr (eV/atom) of the Fe–Mn–Al system.
.
In Table 1, for these Fe3−xMnxAl alloys, the cohesive energy varies from −7.981 eV/atom to −8.190 eV/atom, and the formation enthalpy varies from −0.127 eV/atom to −0.217 eV/atom. We can find that Fe3Al is the most stable from the point view of cohesive energy. But thermodynamic stability is decided by the formation enthalpy of the compound. Lower negative formation enthalpy implies much more stable crystal structure. Figure 2 shows the calculated cohesive energy and formation enthalpy with Mn-content change curves. We can see that the cohesive energy and formation enthalpy increase with Mn atom content increasing. Comparing these formation enthalpies, we can find that the formation enthalpy of Fe3Al is the lowest. Therefore, we believe that Fe3Al is the most stable in the Fe3−xMnxAl system.
Fig. 2. (color online) Variations of cohesive energy and formation enthalpy of Fe3−xMnxAl system.
3.2. Electronic structures
In this section, in order to analyze the chemical bonding characteristics and electronic structures of the Fe3−xMnxAl system, we report our calculations of the total density of states (TDOS), partial density of states (PDOS), and electron density distribution maps. The calculated electronic structures are shown in Figs. 3 and 4.
Fig. 3. (color online) Calculated total density of states and partial density of states for Fe and Fe3−xMnxAl system. Dashed lines represent the Fermi level.
Fig. 4. (color online) (a), (c), (e), (g), (i), (k), (m) Total electron density distribution contours and (b), (d), (f), (h), (j), (l), (n) electron density difference distribution contours for Fe3−xMnxAl system.
The calculated partial density of states and total density of states of the Fe3−xMnxAl system are shown in Fig. 3. All of these Fe–Mn–Al alloys have metallic features because the density of states is non-zero at the Fermi level. The nature of the magnetic characteristics can be understood from the spinpolarized total density of states. Generally speaking, the down and up states are almost symmetric, but comparing the down and up states, we can find that these alloys have magnetic characteristics that are due to the dissymmetry of the down and up states. Fe and Mn are transition metals, for which we need to consider only the role of the outermost electrons. Through the states density of Fe, we can find that it is affected by the Fe-d bands primarily. From Fig. 3, we can find that near the Fermi level in the Fe–Mn–Al alloys, the up and down density of states curves are divided mainly into three sharp peaks. These peaks are primarily determined by the Fe-d and Mn-d states. For Fe3Al, the band range from −5 eV to 3 eV is contributed mainly by the Al-p bands and Fe-d bands. For Mn3Al, the band range from −5 eV to 3 eV is decided mainly by the Al-p bands and Mn-d bands. For other Fe–Mn–Al alloys, the band range from −5 eV to 3 eV is determined mainly by the Al-p bands, Fe-d bands, and Mn-d bands. Near the Fermi level, the TDOS of Fe3Al and Mn3Al are composed mainly of the small Al-d states hybridized with the Fe-d states and Mn-d states, respectively. The TDOS of the other Fe–Mn–Al alloys mainly shows hybridization among the small Al-d states, Fe-d states, and Mn-d states. Additionally, the curves of the PDOS of the Fe3Al and Mn3Al indicate that the Fermi surface is primarily from the contribution of Fe-d states and Mn-d states, respectively, but they also involve small Al-p states. The curves of PDOS of the other Fe–Mn–Al alloys show that the Fermi surface is primarily from the contribution of Fe-d states and Mn-d states, but also involves small Al-p states. From what has been discussed above, we may safely draw the conclusion that the bonding features of the Fe3−xMnxAl system are combinations of covalent bonds and metallic bonds, which may lead to good electronic conductivity, relatively high melting points, and hardness.
The calculated total electron density distribution maps and electron density difference distribution maps of the Fe3−xMnxAl system are shown in Fig. 4. From Figs. 4(a), 4(c), 4(e), 4(g), 4(i), 4(k), and 4(m), we can see that all the Fe, Mn, and Al of core regions have smaller interstitial areas and larger values. Through these pictures, we can see the Fe, Mn, and Al atoms’ covalent bonds features clearly. From these total electron density distribution maps, we can find that the elongated contours along the Al–Fe, Al–Mn, and Fe–Mn bond axes indicate existing covalent interactions. More details about chemical bonding characteristics can be found from the electron density difference distribution in Figs. 4(b), 4(d), 4(f), 4(h), 4(j), 4(l), and 4(n). Some electrons are delocalized and distributed through the circular map in the area among Al, Mn, and Fe atoms, which collectively implies the features of covalent bonding and metallic bonding. The electron areas are actually situated around the Mn, Fe, and Al atoms in the blue color, which indicates that covalent bonds exist between Mn, Fe, and Al atoms. Fe2MnAl, Fe2.5Mn0.5Al, and FeAl3 alloys have the covalent bonding behavior among the Al and Fe atoms, Mn3Al has the covalent bonding behavior among the Al and Mn atoms, Fe0.5Mn2.5Al, FeMn2Al, and Fe1.5Mn1.5Al alloys have the covalent bonding behavior among the Al, Fe, and Mn atoms. Fe2MnAl and Fe2.5Mn0.5Al alloys have the metallic bonding behavior among the Fe atoms. FeMn2Al and Fe1.5Mn1.5Al alloys have the metallic bonding behavior among the Fe and Mn atoms. Fe0.5Mn2.5Al alloys have the metallic bonding behavior among the Mn atoms. Also we can see that the Fe–Mn–Al system exhibits anti-bond effect, such as the Fe–Al anti-bond of Fe3Al, Mn–Al anti-bond of Mn3Al. Other alloys also exhibit Mn–Fe or Al–Mn anti-bond, and so on.
According to the above discussion, these Fe–Mn–Al alloys have magnetic characteristics. The bonding characteristics of these Fe–Mn–Al alloys are combinations of covalent bonds and metallic bonds, also exhibiting an anti-bond effect. The interaction between Fe and Al is mainly covalent bonds. Based on the Fe (1.83), Mn (1.55), and Al (1.61) electronegativity, the electronegativity difference is small, so their covalent interactions are weak. We can conclude that these alloys have weaker covalent bonding effects. These bonding features may lead to relatively high melting points and hardness.
3.3. Mechanical stability, elastic properties, and moduli
The Fe–Mn–Al alloys are potentially high-strength materials, so investigation of mechanical performance is very important. We calculated the elastic constants of the Fe3−xMnxAl system. The results are listed in Table 2. There are three independent elastic constants (C11, C12, and C44) of the Fe3−xMnxAl alloys. From the angle of mechanical properties, we further investigated the stability of the Fe3−xMnxAl system. Here, we discuss the formulas of mechanical stability criteria by the elastic moduli of crystal structure. The mechanical stability conditions can be defined as follows for the cubic system:[29]
Table 2.
Table 2.
Table 2.
Calculated elastic constants (Cij), bulk modulus, shear modulus, Young’s modulus, hardness, B/G, and Poisson’s ratio (σ) of Fe–Mn–Al alloys; the units are GPa.
Calculated elastic constants (Cij), bulk modulus, shear modulus, Young’s modulus, hardness, B/G, and Poisson’s ratio (σ) of Fe–Mn–Al alloys; the units are GPa.
.
From Table 2, we can clearly find that these Fe–Mn–Al alloys are mechanically stable, because the above criteria are satisfied. After the elastic constants are obtained, the bulk modulus (B), Poisson’s ratio (σ), Young’s modulus (E), and shear modulus (G) of polycrystalline crystal can be obtained by the Voigt–Reuss–Hill (VRH) approximation[30,31]
Here, the subscripts R and V indicate the Reuss and Voigt averages. BVRH, BV, andBR are the bulk moduli calculated by Voigt–Reuss–Hill, Voigt, and Reuss approximation methods, respectively. GVRH, GV, and GR are the shear moduli calculated within Voigt–Reuss–Hill, Voigt, and Reuss approximation methods, respectively.
The results of calculations and experiments are listed in Table 2, and our calculated results are in good agreement with the literature.[7,21,32–36] The slight deviations may come from the different calculation methods and experimental conditions. As seen from Table 2, Fe1.5Mn1.5Al has the largest C11, 318.9 GPa, which shows that Fe1.5Mn1.5Al is highly incompressible under uniaxial stress along the crystallographic x axis (ε11). C44 represents the shear modulus on the (100) crystal planes. As seen from Table 2, Mn3Al has the smallest C44 and Fe3Al has the largest C44 in the Fe3−xMnxAl system, with values of 124.4 GPa and 151.0 GPa, respectively. The bulk modulus B, shear modulus G, Poisson’s ratio σ, and Young’s modulus E were calculated using Eqs. (5)– (10) and are listed in Table 3. As we know, the bulk modulus reflects the compressibility of the solid under hydrostatic pressure. In the Fe3−xMnxAl system, the bulk moduli are 233.2 GPa, 234.5 GPa, 233.2 GPa, 229.5 GPa, 227.1 GPa, 223.8 GPa, and 225.0 GPa for Fe3Al, Fe2.5Mn0.5Al, Fe2MnAl, Fe1.5Mn1.5Al, FeMn2Al, Fe0.5Mn2.5Al, and Mn3Al, respectively. The shear modulus is resistance to deformations upon shear stress, and the values are 87.9 GPa, 91.1 GPa, 91.5 GPa, 98.8 GPa, 88.4 GPa, 75.5 GPa, and 89.1 GPa for Fe3Al, Fe2.5Mn0.5Al, Fe2MnAl, Fe1.5Mn1.5Al, FeMn2Al, Fe0.5Mn2.5Al, and Mn3Al, respectively. Generally speaking, the greater the shear modulus, the higher the hardness of the compound. Young’s modulus offers a ruler of the stiffness of solid materials. The greater Young’s modulus is, the larger the stiffness of the materials is. As seen from Table 2, Fe1.5Mn1.5Al has the largest Young’s modulus and shear modulus with values of 259.2 GPa and 98.8 GPa, respectively, which indicates that Fe1.5Mn1.5Al may have the largest hardness in the Fe3−xMnxAl system. Poisson’s ratio can reflect the stability of the crystal against shear stress. In this work, we calculated Poisson’s ratios of the Fe3−xMnxAl system and the values range from 0.312 to 0.348. If Poisson’s ratio is close to 0.3, the compound has strong metallic bonding features. Otherwise, Poisson’s ratios deviate from 0.3, which shows that these alloys have covalent bonding. The value of B/G is usually applied to indicate a compound’s brittleness or ductility.[37] The material has the ductile characteristics when B/G ratio is greater than 1.75; otherwise, the material is brittle. In this work, the values clearly imply that all Fe–Mn–Al alloys are ductile. The Vickers hardness (HV) of the Fe3−xMnxAl system is forecast by an empirical model that gives better results for anisotropic structures. The calculation formula is[38]
(11)
Here, k denotes the Pugh’s modulus ratio (k = G/B). The calculated results for the Fe3−xMnxAl system are listed in Table 2. Fe1.5Mn1.5Al has the largest hardness, 7.96 GPa. And Fe0.5Mn2.5Al has the smallest hardness, 4.04 GPa.
Table 3.
Table 3.
Table 3.
Calculated universal anisotropic index (AU), anisotropy (AB and AG), and shear anisotropic factors (A1, A2, A3) of Fe–Mn–Al alloys.
.
Species
AU
AB
AG
A1 = A2 = A3
Fe3Al
2.59
0
0.205
3.91
Fe2.5Mn0.5Al
1.68
0
0.144
3.12
Fe2MnAl
1.23
0
0.110
2.59
Fe1.5Mn1.5Al
0.51
0
0.049
1.90
FeMn2Al
1.15
0
0.103
2.50
Fe0.5Mn2.5Al
2.58
0
0.205
3.90
Mn3Al
0.88
0
0.081
2.30
Table 3.
Calculated universal anisotropic index (AU), anisotropy (AB and AG), and shear anisotropic factors (A1, A2, A3) of Fe–Mn–Al alloys.
.
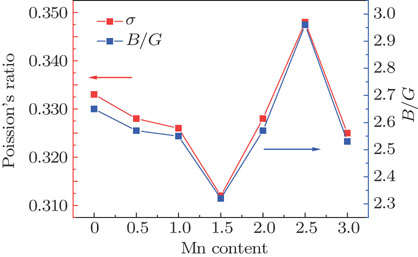
In order to further depict the mechanical properties of the Fe3−xMnxAl system, some elastic moduli with change to Mn atom content are curve listed in Figs. 5–7. From Fig. 5, we can find that the Young’s modulus and C11−C12 have similar variation tendencies. As can be seen from Fig. 6, the bulk modulus and C44 have similar variation tendencies, but the relationship among shear modulus, bulk modulus, and C44 cannot be observed clearly. From Fig. 7, we can see that Poisson’s ratio and B/G have the same change trend.
Fig. 6. (color online) Variations of the bulk modulus, shear modulus, and C44 of Fe3−xMnxAl system. Note that the shear modulus is magnified by 2 times.
Fig. 7. (color online) Variations of Poisson’s ratio and B/G of Fe3−xMnxAl system.
3.4. Mechanical anisotropy
All of the single crystals are actually anisotropic, therefore, a suitable parameter describing the anisotropic degree is necessary. Meanwhile, the appearance of micro cracks in materials is always connected with the anisotropy. The micro cracks often form in materials and harm the performance of the materials. Fe–Mn–Al alloys are potential high-strength materials, so the anisotropic mechanical properties of these alloys are investigated carefully. We calculated several anisotropic indexes, including the anisotropy index (AB and AG), the shear anisotropic factors (A1, A2, and A3), and universal anisotropic index (AU). The equations are as follows:[39]
(12)
(13)
(14)
(15)
where GV, GR, BV, and BR are the shear modulus and bulk modulus estimated within Voigt and Reuss methods, respectively. If all indexes are zero, the crystal structure is isotropic. And deviations from zero show anisotropy. A greater anisotropy index (AU) indicates higher mechanical anisotropic properties. The calculated results are listed in Table 3. The calculated AB of all alloys is zero because the bulk moduli are the same by using the Voigt or Reuss methods. AG has a low value for these alloys, which shows that these alloys have a weak anisotropy in shear modulus. The universal anisotropic index AU is considered to be a better indicator for mechanical anisotropic properties of these alloys. We can clearly find that Fe1.5Mn1.5Al has the smallest AU among the Fe–Mn–Al alloys. The AU value for Fe3Al is larger than that of the other Fe–Mn–Al alloys, indicating that Fe3Al has the largest elastic anisotropy among the Fe–Mn–Al alloys. Generally speaking, the larger the value of AU is, the stronger the anisotropy is. So the descending order of anisotropy for Fe–Mn–Al alloys is Fe3Al > Fe0.5Mn2.5Al > Fe2.5Mn0.5Al > Fe2MnAl > FeMn2Al > Mn3Al > Fe1.5Mn1.5Al. AG, A1, A2, and A3 depend on the anisotropy of the shear modulus. A1, A2, and A3 represent the anisotropies of the shear modulus in the (100), (010), and (001) planes, respectively. From Table 3, Fe3Al has the strongest anisotropy of the shear modulus in the (100), (010), and (001) planes, and the A1, A2, and A3 values are 3.91. In the (100), (010), and (001) planes, Fe1.5Mn1.5Al has the weakest anisotropy of the shear modulus among these Fe–Mn–Al alloys, and the A1, A2, and A3 values are 1.90. In addition, from Fig. 8, we can find that AG and AU have the similar trend.
Fig. 8. (color online) Variations of the AG and AU of Fe3−xMnxAl system.
In order to better depict the features of elastic anisotropy, we plotted a curved surface of three dimensions (3D). In this work, the Young’s modulus is plotted at different directions using spherical coordinates. And the directional dependence of Young’s modulus can be given by the following equations for cubic crystal:[40]
Here, l1, l2, and l3 are the directional cosines, and Sij are the elastic compliance constants. In the spherical coordinates, l1 = sin[θ]cos[ψ], l2 = sin[θ]sin[ψ], and l3 = cos[θ]. The angle between θ and ψ is defined on the horn in the spherical coordinates. The 3D curved surface of the Young’s modulus of the Fe3−xMnxAl system is shown in Fig. 9. These graphs represent the size of Young’s modulus along different directions. Obviously, figure 9 shows that these Fe–Mn–Al alloys have a strong anisotropy feature in Young’s modulus. Meanwhile, we can clearly see that these alloys have similar anisotropies; Fe3Al has the strongest anisotropy of Young’s modulus, while Fe1.5Mn1.5Al has the weakest. More details of the anisotropic properties of Young’s modulus on the (001) and (110) planes are illustrated in Fig. 10. On these planes, we can clearly see that the Young’s modulus has a strong directional dependence. At the [100], [010], [001], and [110] directions, we can find that Fe1.5Mn1.5Al shows the maximum Young’s modulus, while Fe0.5Mn2.5Al shows the minimum Young’s modulus. For these alloys, the planar contours on the (001) and (110) planes are not similar to a spherical shape, which indicates that the Young’s modulus of these alloys shows strong anisotropy on the (001) and (110) planes.
Fig. 10. (color online) Planar projections of the Young’s modulus of the Fe3−xMnxAl system on the (a) (001) and (b) (110) crystallographic planes.
3.5. Anisotropic sound velocities and Debye temperature
The sound velocities of transverse and longitudinal modes of the Fe3−xMnxAl system are calculated using the single crystal elastic constants.[41] The sound velocities are determined by the propagation direction and symmetry of the crystal. The pure longitudinal and transverse modes are found in [100], [110], and [001] directions, and the sound propagating modes in other directions are the quasi-longitudinal or quasi-transverse waves. In the principle directions, the acoustic velocities can be calculated using the following relations:[42–44]
Here, vl is the longitudinal sound velocity; vt1 is the first transverse modes; vt2 is the second transverse modes; and ρ is the theoretical density. The calculated anisotropic sound velocities of the Fe3−xMnxAl alloys are listed in Table 4. Obviously, these alloys have large elastic moduli and smaller densities, leading to fast sound velocities.
Table 4.
Table 4.
Table 4.
Anisotropic sound velocities of Fe–Mn–Al alloys; the unit of velocity is m·s−1.
.
Species
[100]
[110]
[001]
vl
vt1
vt2
vl
vt1
vt2
vl
vt1
vt2
Fe3Al
6198.2
4514.0
4514.0
7320.2
3227.6
4514.0
7657.7
3203.8
3203.8
Fe2.5Mn0.5Al
6354.5
4396.6
4396.6
7307.2
3553.5
4396.6
7598.3
3263.8
3263.8
Fe2MnAl
6456.3
4301.2
4301.2
7283.9
3776.1
4301.2
7539.6
3304.5
3304.5
Fe1.5Mn1.5Al
6691.5
4241.0
4241.0
7302.1
4345.7
4241.0
7494.5
3505.8
3505.8
FeMn2Al
6502.1
4303.7
4303.7
7306.0
3852.7
4303.7
7555.0
3334.9
3334.9
Fe0.5Mn2.5Al
6223.0
4326.6
4326.6
7256.8
3093.5
4326.6
7570.1
3070.8
3070.8
Mn3Al
6586.8
4261.5
4261.5
7324.5
3974.3
4261.5
7554.4
3364.3
3364.3
Table 4.
Anisotropic sound velocities of Fe–Mn–Al alloys; the unit of velocity is m·s−1.
.
The Debye temperature is a fundamental parameter and is related to some physical properties of materials, such as specific heat, elastic constants, and melting point.[45] It is obvious that the Debye temperature and sound velocity can be used to estimate the chemical thermal properties and chemical bond behavior of compounds. The average sound velocity (vm) and Debye temperature (ΘD) can be calculated using the following equations:[46]
(18)
(19)
(20)
(21)
where ΘD is the Debye temperature; kB and h are the Boltzmann and Planck constants, respectively; NA is the Avogadro constant; n is the total number of atoms per formula; M is the molecular weight per formula; vs is the transverse sound velocity; vl is the longitudinal sound velocity; G and B represent the shear modulus and bulk modulus, respectively.[42]
We calculated the sound velocities and Debye temperature of the Fe3−xMnxAl system, and the values are listed in Table 5. In general, the Debye temperature can help understand the thermal properties and chemical bonding features of a compound. For crystal structure, the Debye temperature can reflect the strength of covalent bonding. From Table 5, we can find that Fe1.5Mn1.5Al has the largest ΘD, with the value of 551.1 K. So we conclude that Fe1.5Mn1.5Al has stronger covalent bonds than other FexMn3−xAl alloys. Fe0.5Mn2.5Al has the highest B/G ratio and lowest ΘD, at 487.2 K, which shows the strongest metallic character. More details can be found from Fig. 11. Obviously, the Debye temperature increases from Fe3Al to Fe1.5Mn1.5Al with the increases of Mn content, but the Debye temperature decreases from Fe1.5Mn1.5Al to Fe0.5Mn2.5Al. The hardness and Debye temperature have the same change trend, maybe because the bonding features of these alloys have the same trend effects for hardness and Debye temperature. In addition, the calculated average sound velocities of the Fe3−xMnxAl system are relatively large, because they have large elastic moduli and smaller densities, and the v1 and vs values are concerned with density, shear modulus, and bulk modulus.
Table 5.
Table 5.
Table 5.
Theoretical density (ρ, kg·m−3), longitudinal sound velocity (vl, m·s−1), transverse sound velocity (vs, m·s−1), average sound velocity (vm, m·s−1), and Debye temperature (ΘD, K).
.
Species
ρ
v1
vs
vm
ΘD
Fe3Al
7.41
6876.3
3444.0
3863.2
516.6
Fe2.5Mn0.5Al
7.33
6970.9
3526.5
3953.3
527.0
Fe2MnAl
7.24
7054.2
3555.6
3985.1
529.6
Fe1.5Mn1.5Al
7.12
7121.8
3724.6
4166.5
551.1
FeMn2Al
7.01
7013.4
3550.3
3979.8
524.2
Fe0.5Mn2.5Al
6.92
6848.6
3303.6
3713.5
487.2
Mn3Al
6.85
7084.4
3606.5
4041.4
528.9
Table 5.
Theoretical density (ρ, kg·m−3), longitudinal sound velocity (vl, m·s−1), transverse sound velocity (vs, m·s−1), average sound velocity (vm, m·s−1), and Debye temperature (ΘD, K).
Fig. 11. (color online) Variations of the hardness and Debye temperature of the Fe3−xMnxAl system.
4. Conclusion
The stability, electronic structures, mechanical properties, and Debye temperature of these Fe–Mn–Al alloys were studied by first-principles calculations. The cohesive energy and formation enthalpy illustrate that the alloys are thermodynamically stable. We calculated the electron density distribution maps and density of states. We deduced that the bonding characteristics of the Fe–Mn–Al alloys are dominated by covalent bonds and metallic bonds. The elastic constants of these Fe3−xMnxAl alloys reflect the mechanical stability conditions, which indicate that the Fe–Mn–Al alloys are thermodynamically stable. We calculated the elastic constants (Cij), shear modulus (G), Poisson’s ratio (σ), bulk modulus (B), and Young’s modulus (E) by Voigt–Reuss–Hill (VRH) approximation. Fe1.5Mn1.5Al has the largest Young’s modulus and shear modulus. We can speculate that Fe1.5Mn1.5Al is the hardest Fe3−xMnxAl species. We calculated several anisotropy indexes to estimate the mechanical anisotropy, which together, indicate that Fe3Al has the strongest anisotropy. Young’s moduli of these Fe–Mn–Al alloys are plotted to show the anisotropy by a 3D surface contour. These alloys have strong anisotropy characteristics in Young’s modulus. The anisotropic sound velocities and Debye temperature for these alloys were also calculated. Fe1.5Mn1.5Al has the largest Debye temperature, 551.1 K, which shows that Fe1.5Mn1.5Al has stronger covalent bond features than the other alloys.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
























